Plant material, transformation and generation of transgenes
In this study, we used the variety Agria (the predominant cultivated variety in Iran) which based on their previously classed as both frost and drought sensitive [42, 43]. Agria seed and tuber was obtained from Seed and Plant Improvement Institute (SPII), Karaj. To remove possible viral infections, the seeds were treated following a procedure used by Kaiser [44]. Then, the seed surface was disinfected by immersion in 0.5% hypochlorite sodium (NaClO) for 10 min before being rinsed and washed thoroughly in sterile distilled water thrice. The seeds were rinsed in a laminar airflow cabinet and for further use placed on an aseptic blotting paper in the disinfected dishes. To encourage the germination of potato seed sprouts, the seeds were placed in darkness at 25±2°C and 80% relative humidity in a growth chamber. To activate the vir gene, when the sprouts grew about 4 to 6 mm in length, the Agrobacterium tumefaciens cell pellet was suspended in an autoclaved MS medium (0.5 X) containing Acetosyringone (50 µM). Cell suspensions were adjusted to an optical density of 1 at 600 nm (OD600 =1). Then, the suspension was drawn into a 1ml insulin syringe, and the sprouts (slightly below the tip) received injections under a binocular microscope without rupturing. There were three control treatments planned as a negative control, the injected sprouts with MS medium and without Acetosyringone content as well as non-injected sprouts. The injections were renewed three times at 24-hour intervals. Then, the seeds were stored at 25±2°C in darkness at 80% relative humidity for 48 h in the growth chamber. The seeds were sown in pots containing autoclaved peat moss and were grown in a greenhouse. In total, 50 replicates were set up for each injected (with Acetosyringone and without Acetosyringone treatments) and control treatments (Figure 1).
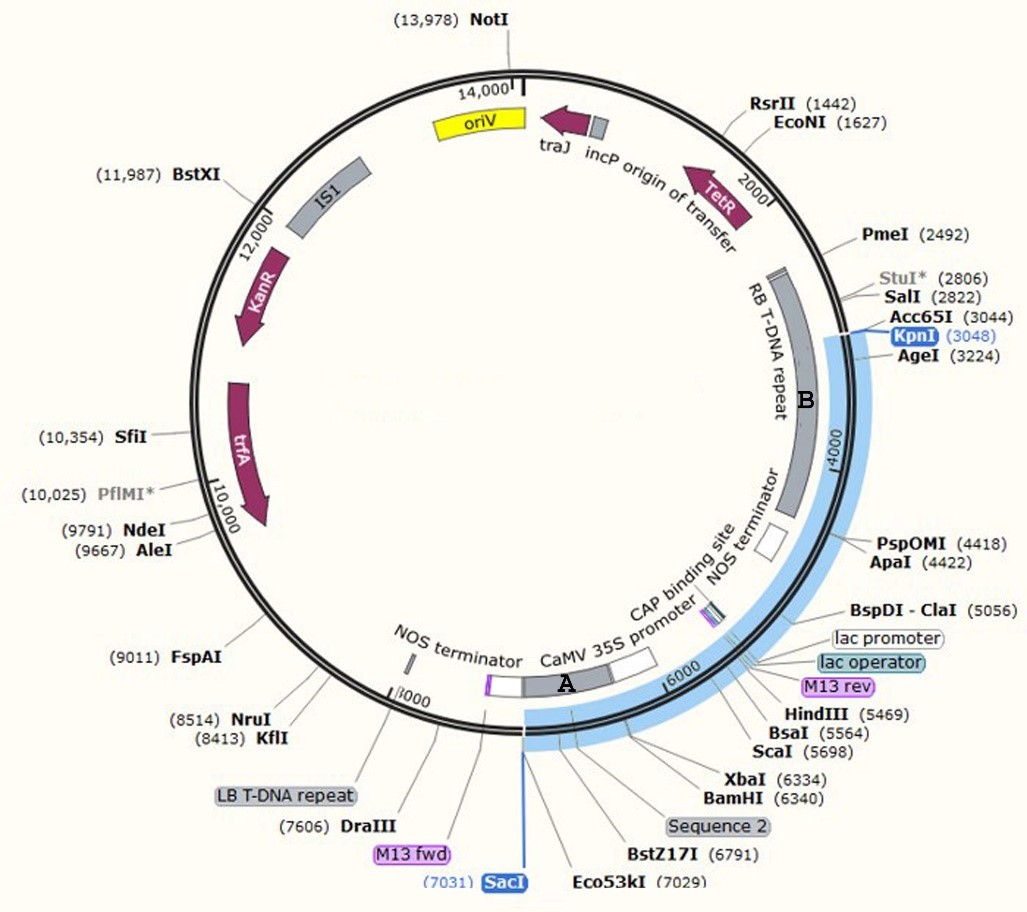
Plasmid construct and Agrobacterium strain
The hva1 gene sequence originated from Hordeum vulgare. It was obtained from the gene database of NCBI (X78205.1 access number including 1804nt). The sequence was optimized based on the potato codon-usage preference table using Gene Designer Gene2 software. Then, the Kozak and His-tag sequences were designed upstream of the hva1 coding sequence, and the KDEL (endoplasmic reticulum transmitter facilitator) was added to downstream hva1 coded protein for proper protein folding. A sequence-specific primer for the gene coupling was designed and synthesized via Vector NTI software. Also, at the 5’ head, each of the two nucleotides was designed as an enzyme sitting site. For a long-term reproduction and preservation of the structure, it was initially shifted to the E. coli PUC57 vector and cloned. Then, the plasmid was extracted from E. coli, and the hva1-harboring plasmid PUC57 vector was performed in association with SacI and BamHI enzymes, and the hva1-containing structure was released. For the structural transformation, a modified pBI121 binary plasmid expression vector served as a carrier. The vector consisted of a 1.4 kb DNA fragment containing 5-enolpyruvylshikimate-3-phosphate synthase (EPSPS; A Glyphosate-resistant gene) as a plant-selection-marker. The gene cassette was cloned in the plant expression vector, being under the expressional control of an enhanced 35S promoter from the cauliflower mosaic virus (CaMV). From then on, the expression carrier was called hva1-pBI121 (Supplementary file 1).
SacI and KpnI enzymes were applied to confirm the presence of hva1 in the structure. The enzymes cut the plasmid at the relevant designed restriction sites and relaxed the 10091 and 3938 bp segments, which confirmed the presence of EPSPS in the complex. Also, colony PCR of the structure was carried out to confirm the presence of hva1 in the complex. Furthermore, to confirm the existence of hva1 in the vector, enzymatic digestion was performed using SacI and KpnI enzymes (the KpnI cleavage site is in the middle of the EPSPS gene, which is not naturally present in pBI and confirms the presence of EPSPS in the structure).
Agrobacterium tumefaciens strain C58 contained the binary vector hva1-pBI121. This strain was used as a vector-transferring agent. The bacterium was streaked onto LA medium (10gL-1 tryptone, 5 gL-1 yeast exact and 5 gL-1 NaCl, and Agar 7 gL-1), which had been supplemented with 50 mg Kanamycin and 50mg Rifampicin. Then, an individual colony was randomly sampled from the culture and re-cultured in a modified LB medium (containing 50 mgL-1 Rifampicin 50 mgL-1 Kanamycin). Then, the culture was incubated in the growth chamber for 48h at 28°C ±1 on a rotary (at 135 rpm) in dark conditions to form homologous colonies (OD600=0.3). The explants were treated with A. tumefaciens cell pellets in an autoclaved MS medium (0.5 X) containing Acetosyringone (50 µM). This was to achieve a higher transformation efficiency. To study the effects of Acetosyringone content on the efficacy of the transformation, the explants were also treated by A. tumefaciens cell pellets in MS medium with no Acetosyringone, which served as the control.
Glyphosate lethal concentration and Glyphosate screening of putative transformants
To evaluate the Glyphosate lethal concentration in transgenic and non-transgenic potatoes, the experiment was designed in two groups of transgenic and non-transgenic potato plants by five Glyphosate dosages (0%, 0.25%, 0.5%, 1%, and 2% v/v) in four replicates for each treatment. Potato tubers were placed in pots and stored in the greenhouse at 20–30 °C while receiving 16/8 hr day/night light. The plants were watered every seven days. When the plantlets had grown 15-30 cm tall, they were sprayed thoroughly with the Glyphosate solution in ddH2O. Morphological changes of the treatment were considered in comparison with the control. The changes were monitored at intervals of one, two, three, and four weeks after the treatment. Necrotic lesions were considered and the damage was reported in percentages (Compared to healthy tissues). Statistical significance was determined by one-way analysis of variance (ANOVAs) and by Tukey’s Multiple Comparison test (P≤0.05).
In this study, we used the EPSPS as a marker to distinguish transformants from non-transformants. Also, for eliminating chimeric plants, the two-stage selection was performed with 1 and 2 % V/V Glyphosate concentration respectively and monitored for the foliage tolerate. The transformants, which tolerated the Glyphosate treatments and continued to survive, were selected for further experiments. Non-transformed plants and the control group either achieved no growth or died.
PCR- mediated confirmation of putative transformants
The genomic DNA was extracted carefully from the leaves of treated plants according to the method of Cetyl Trimethyl Ammonium Bromide (CTAB) extraction, as modified by Gawel and Jarret (1991). Purified DNA was stored in TE buffer at -20°C for further use. The standard Polymerase Chain Reaction (PCR) for hva1 was carried out in a 25µl reaction mixture containing purified DNA 100ng, 250 µM for each dNTP, 10 pM of hva1 specific designed primers (Forward primer 5’-ACACTTCTGATACACCTTTC-3’ and Reverse primer 5’-GTTACCATTTCCCACACC-3’), 2mM MgCl2, 1.25 U Taq DNA polymerase and Taq buffer. The reaction mixture in the tubes was placed in a programmable thermal cycler model MJ research PTC-200. The thermal cycler was programmed as follows: setting an initial denaturation at 94°C for 5 min, 30 amplification cycles (denaturalizing at 94°C for 45 s, annealing at 60°C for 30s and extension at 72°C for 20 s). Then, a final extension was performed at 72°C for 10 min. Also, the PCR reaction was performed on 20 µl reaction mixtures containing 100ng purified DNA, 250 µM of each dNTP, 10 pM EPSPS specific primers (while the sequences of the primers were as follows: forward primer 5’- TTGGTTGTCAGAGGTAGA-3’ and Reverse primer 5’- AGCAGCCTTAGTATCAGA-3’), and 0.75 U Taq DNA polymerase for amplification of EPSPS sequence and verification. The PCR reaction program began at 94°C for 4 min to allow an initial denaturation, followed by 30 amplification cycles (for 45s to allow denaturation at 94°C, 30s of annealing at 60°C and 20s of extension at 72°C). A final extension was applied at 72°C for 30s. Also, a PCR was carried out using the virG specific primers (C58F; 5’-TTACGCAGCAGGTCTCAT-3’ and C58R; 5’-CGAAGGATAGTGGGATTGTG-3’) on putative transgenic lines. This was to detect contamination with Agrobacterium and to confirm the integration of the structure into the plant genome, exactly one month after injection. All PCR-tested samples were separated on 2% TAE-buffered agarose gels, stained with ethidium bromide (0.5 µg/ml) for 20 min, and visualized with UV illumination. No specific fragment was amplified on putative transgenic lines, which shows that the lines weren't contaminated with Agrobacterium (GeneRuler DNA Ladder Mix).
Semi-Quantitative Reverse Transcription (RT)-PCR
The total RNA was extracted from the leaves of both positive PCR-tested transformants and control plants by an RNA extraction kit (DENAzist Ltd. IRAN). The contaminating DNA was removed by a DNaseI DNase Kit. Then, the elimination of DNA contamination was confirmed by the absence of bands on the gel. The transgene expression was confirmed by carrying out RT-PCR. The DNase-treated extracted RNA was converted initially to complementary DNA (cDNA) using 1µg total RNA in a 20 µl RT-reaction containing oligo dT as a primer and RevertAid enzyme. The reaction mixture was subjected to denaturation at 65 °C for 5 min before cooling down to 37°C. Then, 40 U of M-MuLV entered the reaction mixture allowing an extension at 37°C for 1 h, but which later was stopped by being heated at 70°C for 10min. Subsequently, 20µl of the cDNA was used as a template for an exponential amplification reaction using nptII primers (Forward, 5’-AGATCCCGTGGGCGAAGAACT-3’ and Reverse, 5’-GGATCGTTTCGCATGATTGAA-3’). The reaction mixture contained 1× PCR buffer, 50 ng cDNA template, 1 pmol of each primer and 2 U of Taq DNA polymerase. Thirty PCR amplification reaction cycles (causing denaturation at 94°C for 30s, annealing at 65°C for 30s and extension at 72°C for 1 min) were performed after an initial denaturation condition at 94°C for 5 min, followed by a final elongation at 72°C for 8 min. Also, RT-PCR for hva1 and EPSPS was carried out when appropriate. The PCR products were separated by 1% agarose gel electrophoresis.
Crude protein extraction and Enzyme-linked immunosorbent assay (ELISA) test
At this stage of the experiment, all the positive PCR-tested transformants were considered for extracting total crude protein from their young leaves. The samples in four replicates were ground under liquid nitrogen and the powder was suspended in 1:1 phosphate buffer (100 mM, pH=7) w/v. Then, the supernatant was prepared by centrifuging at 12000 ×g for 10 min at 4°C. The protein concentration was determined by the Bradford method (Kruger 2009) and the proteins were stored at -20°C before use. ELISA was conducted with anti-His-tag polyclonal IgG according to the manufacturer’s instructions (Biolegend, USA). After 45 min of incubation with in the substrate, the absorbance was measured at 450 nm using an ELISA reader. Two controls (Bovine serum albumin (BSA) and the non-treated plant samples) were set in this experiment. Four replicates were set for each treatment. The results were confirmed by repeating the experiment thrice. The differences between the control group and the test samples were analyzed by one-way analysis of variance (ANOVAs) and by Tukey’s Multiple Comparison Test. A P-value of less than 0.05 was set as statistical significance.
Effect of the integrated genetic material expression on plant growth and tuberization
The expression of integrated genetic material may affect the plant growth and tuber formation. These were examined by comparing the phenotypes of the control plants with the transgenic plants grown in the greenhouse for ten weeks. The plant weight, height, number of potato tubers and physical appearance were considered. The experiment was a completely randomized design (CRD) in eight replicates for each transgenic and non-treated plant. Statistical significance was determined using the one-way analysis of variance (ANOVAs) and by Tukey’s Multiple Comparison test (P≤0.05).
Stability of the transgene in tubers
The stability and expression of the transgene in the tubers were also considered. The results were analyzed based on the presence or absence of amplified PCR products (the hva1 and EPSPS producing bond) and ELISA with anti-His-tag polyclonal IgG in the PCR-positive transgene being generated in tubers and shoots.
Whole plant freezing stress treatment
To determine how tolerant transgenic plants are to freezing stress, eight-week-old plants were placed in a growth chamber at 25±1°C and at 100µmol/m2s light intensity. Then, they were exposed to -4°C for 12h. Following the freezing treatment, they were returned to natural conditions for two weeks in order to recover. Transgenic potato tolerance was assessed by analyzing plant survival compared to non-transgenic potatoes as the control. For each transgenic and non-transgenic potato group, six replicates were grown in a growth chamber and were exposed to freezing temperatures.
{kind=link}