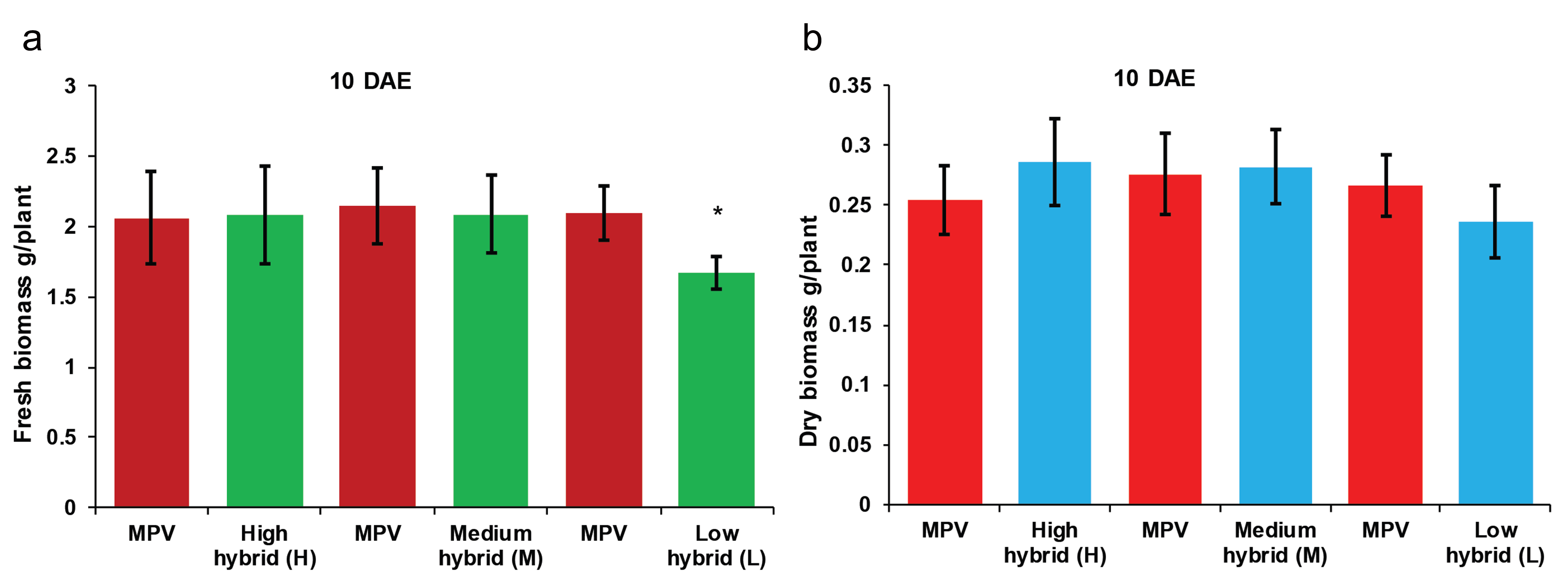
Biomass heterosis exist in contrasting hybrids at the seedling stage
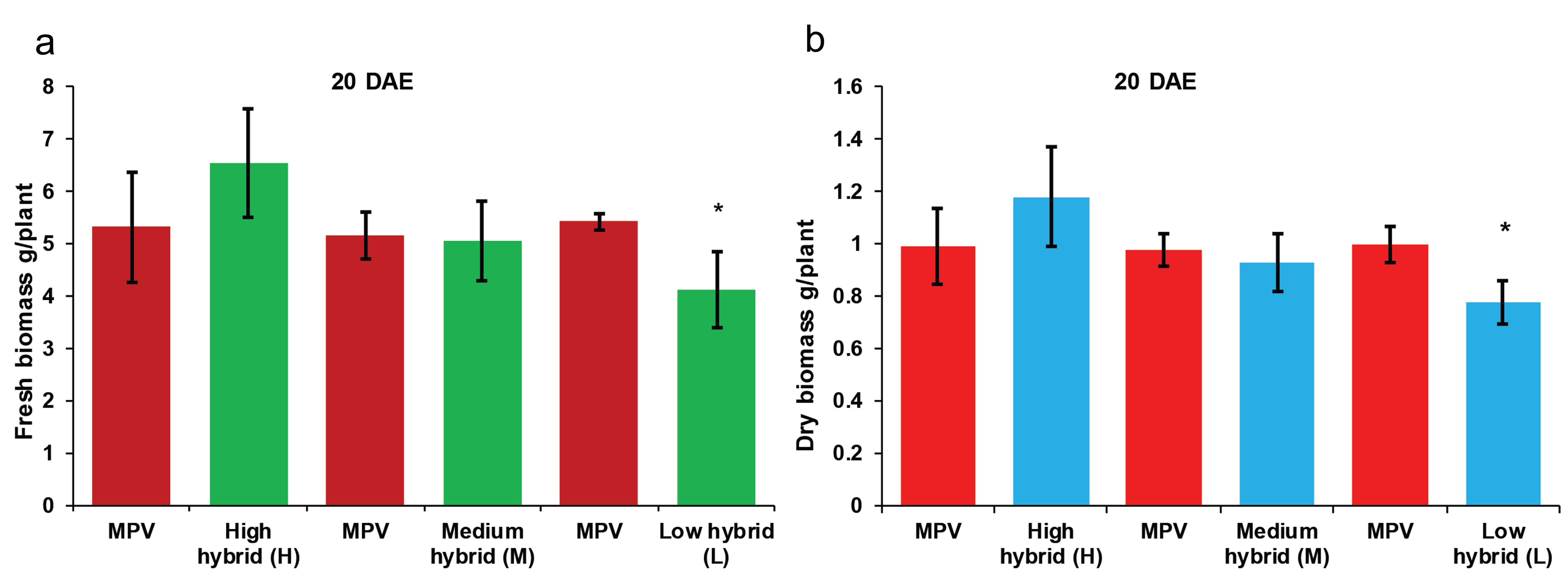
Three contrasting hybrids having high (H), medium (M), and low (L) parent heterosis in yield traits together with their four inbred parents (denoted as A, B, C, and D) were used to investigate the early biomass vigor in the field. We observed high and medium hybrids did not show statistically significant difference in fresh biomass (g/plant) and dry biomass (g/plant) relative to their mid-parent values (MPV) at 10 days after emergence of seedling (DAE) and 20 DAE (Fig. S1 and Fig. S2). However, high hybrid showed highly significant difference in fresh biomass compared with its MPV at 30 DAE (Fig. 1). According to the results, low hybrid showed significant difference in fresh biomass compared with its MPV at 10 DAE (Fig. S1). Similar results were obtained at 20 DAE (Fig. S2) and 30 DAE (Fig. 1). High hybrid showed more biomass accumulation and low hybrid showed decreased biomass accumulation compared to their parents. However, medium hybrid did not show any difference in biomass accumulation compared with both parents. These results indicate biomass heterosis is established in hybrids compared with their parents during early seedling growth.
Transcriptome sequencing, mapping and global expressions in root and leaf
Three contrasting F1 hybrids and their four inbred parents were used to perform RNA sequencing in this study. Root and leaf tissues of the same plant with three biological replicates were used to build 42 cDNA libraries for RNA Illumina sequencing. The brief detail of sequencing, mapping and gene expressions of the individual library is presented in additional file 1. The overall sequencing means of valid reads was 98.8 % with a value of 90% exon region mapping. The value of Q20% was 99.6% in our sequencing results. In the root of A, B, C, D, H, M, and L genotypes, mean of valid reads was approximately 51, 54, 50, 52, 43, 46, and 45 million, respectively (Table 1). The mean of valid reads in the leaf of A, B, C, D, H, M, and L genotypes was ~50, 49, 42, 44, 45, 43, and 45 million, respectively. On an average, more than 89% valid reads were mapped to G. hirsutum TM-1 reference genome in this sequencing. However, the mapped percentage in root samples was lower than leaf. The mean of multi-mapped and splice reads was more than 28% and 31%, respectively. In this study, the root tissue of each genotype showed higher number of total expressed genes compared to leaf. For example, approximately 77 thousand genes were expressed in root of A, while it was ~ 75 thousand in leaf (Additional file 1). We also found total numbers of expressed genes in both tissues of F1 hybrids were higher than the parents (Additional file 1).
Differentially expressed genes in root and leaf transcriptome
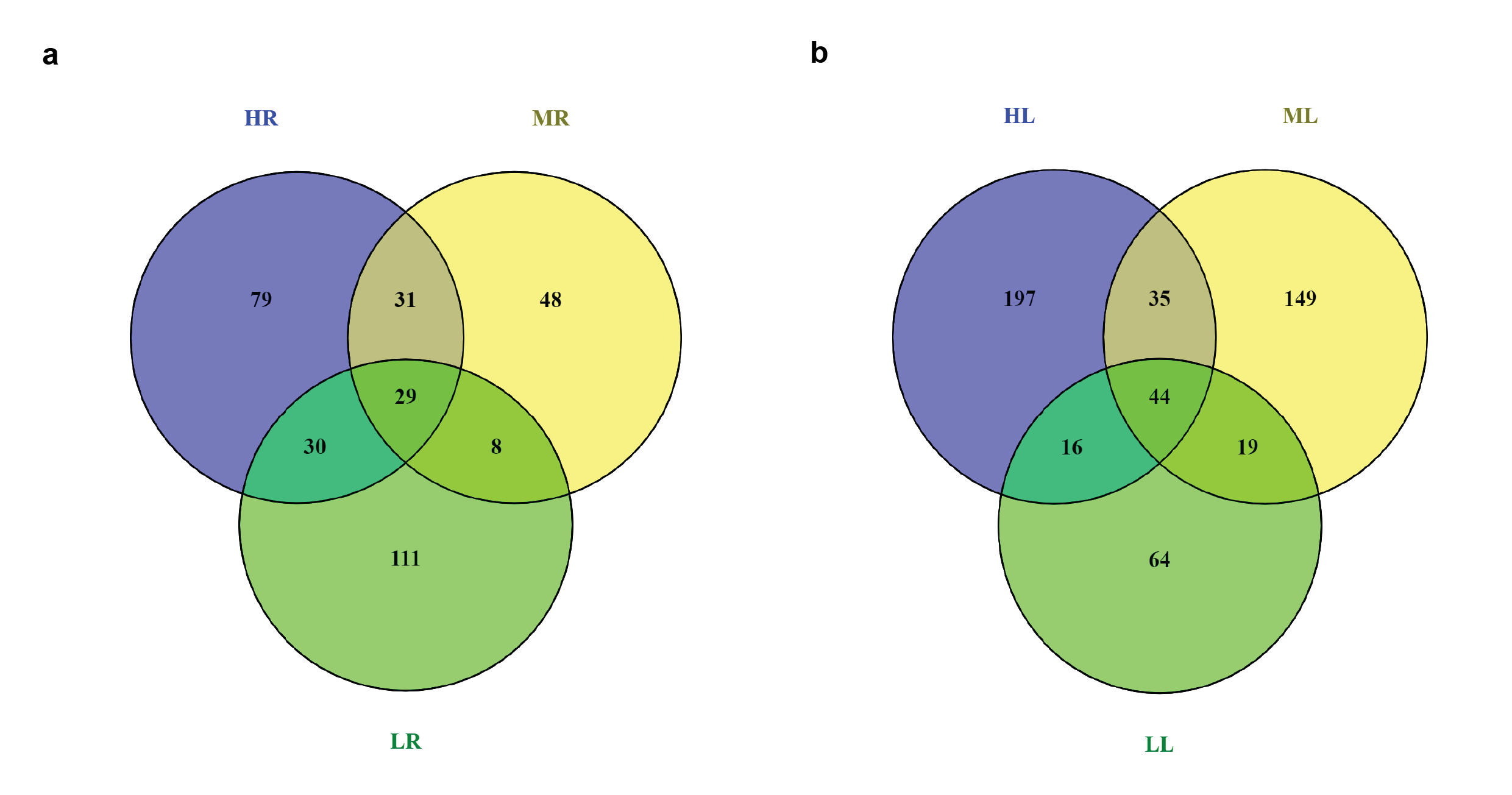
Here, we analyzed the dynamic changes of root and leaf transcriptome in all F1 hybrids and their inbred parents. The expression levels significantly different at p ≤ 0.05 and with log2 (fold change) >1 or log2 (fold change) < -1 designated as differentially expressed genes in each hybrid parent triad. The total number of DEGs (Up + down) among each hybrid parent triad is represented in Fig. 2a. The comparative analysis in the root of hybrid (H) compared with both parents revealed H with maternal parent (A) and paternal parent (B) respectively showed 2917 and 2828 total number of DEGs (Figure 2a: Additional file 2). The comparison of both parents ARvsBR showed 1154 total number of DEGs. In the leaf transcriptome of H, the comparison of ALvsHL showed 3807 total number of DEGs, whereas the comparison of BLvsHL showed a total of 2797 DEGs (Fig. 2a: Additional file 3). Furthermore, the comparison of both parents ALvsBL showed 1013 total number of DEGs. Distribution of DEGs in H-hybrid revealed major portion of genes was unique and less was common in each tissue (Fig. 2b and c). For instance, 2631 genes in roots and 2993 in leaf were unique in the combination of H with A. Unique DEGs in comparison of B and H was 2648 in root and 1974 in leaf. A large portion of differential gene expression is probably major determinant of better performance in high hybrid.
The root transcriptome of medium hybrid (M) revealed comparison of M with maternal parent (A) and paternal parent (C), respectively showed 2577 and 2144 total number of DEGs (Fig. 2a: Additional file 4). It was observed the comparison of ALvsML had 3910 total number of DEGs in the leaf of M hybrid, whereas the comparison of CLvsML showed 2170 total number of DEGs (Fig. 2a: Additional file 5). Moreover, the combination of both parents had 1067 DEGs in root and 1586 in leaf. Although results of unique and common DEGs distribution in M hybrid identified more unique but less common genes. However, unique DEGs were more in the comparison of M with A than with C (Fig. 2d and e). The comparative analysis revealed total number of DEGs between low hybrid (L) and its maternal parent (A) was 3580 in the root (Fig. 2a: Additional file 6). The combination of L and its paternal parent (D) had 3215 total number of DEGs. Furthermore, the comparison of ALvsLL and DLvsLL, respectively showed 5827 and 2623 total number of DEGs (Fig. 2a: Additional file 6). Distribution of DEGs exposed that the majority of genes were unique, whereas less was overlap (Fig. 2f and g). For example, A versus L had 2647 unique DEGs in root and 4795 in leaf. Unique DEGs in DvsL were 2413 in root and 1714 in leaf. More interestingly, both parents of low hybrid had higher genetic differences among each other than those of medium and high hybrids. Overall, the result of comparative analysis of DEGs between hybrids and parents revealed that hybrids had different genomic constituent relative to their parents. However, comparison of both parents indicated that they have few genetic differences among them.
F1 hybrids exhibited overdominant gene expressions
Allopolyploids have been found to exhibit expression level dominance [35, 40]. So, to address the magnitude and directionality of expressions in interspecific F1 upland cotton hybrids, DEGs of root and leaf transcriptome were divided into 12 possible groups as described by Rapp et al., [41]. Genes in groups 1-2 showed additive expression. The expression of genes in groups 3-4 was termed as male expression level dominance (M-ELD), wherein the expression of genes in groups 5-6 was named as female expression level dominance (F-ELD). The expression of genes in groups 7-9 called downregulated overdominance, wherein the expression of genes in groups 10-12 was named as upregulated overdominance (Fig. 3a). The result of expression patterns analysis in high hybrid (H) in both tissues detected limited number of genes was additive expressions. Male and female parent like expression groups also had few genes. However, it was found overdominant upregulated (group12) and downregulated (group 8) groups had the highest number of genes in both tissues (Fig. 3b: Additional file 8). The analysis results of medium hybrid (M) also dissected overdominant up and downregulated groups had the highest number of genes in both tissues (Fig 3c: Additional file 9). The analysis result of low hybrid (L) was also similar to H and M hybrids (Fig. 3d: Additional file 10). However, groups of parent-like expression also had many genes in L hybrid as compared to H and M hybrids. The result of expression patterns analysis directed that overdominance at the gene expression level contributes to early biomass vigor in hybrid cotton.
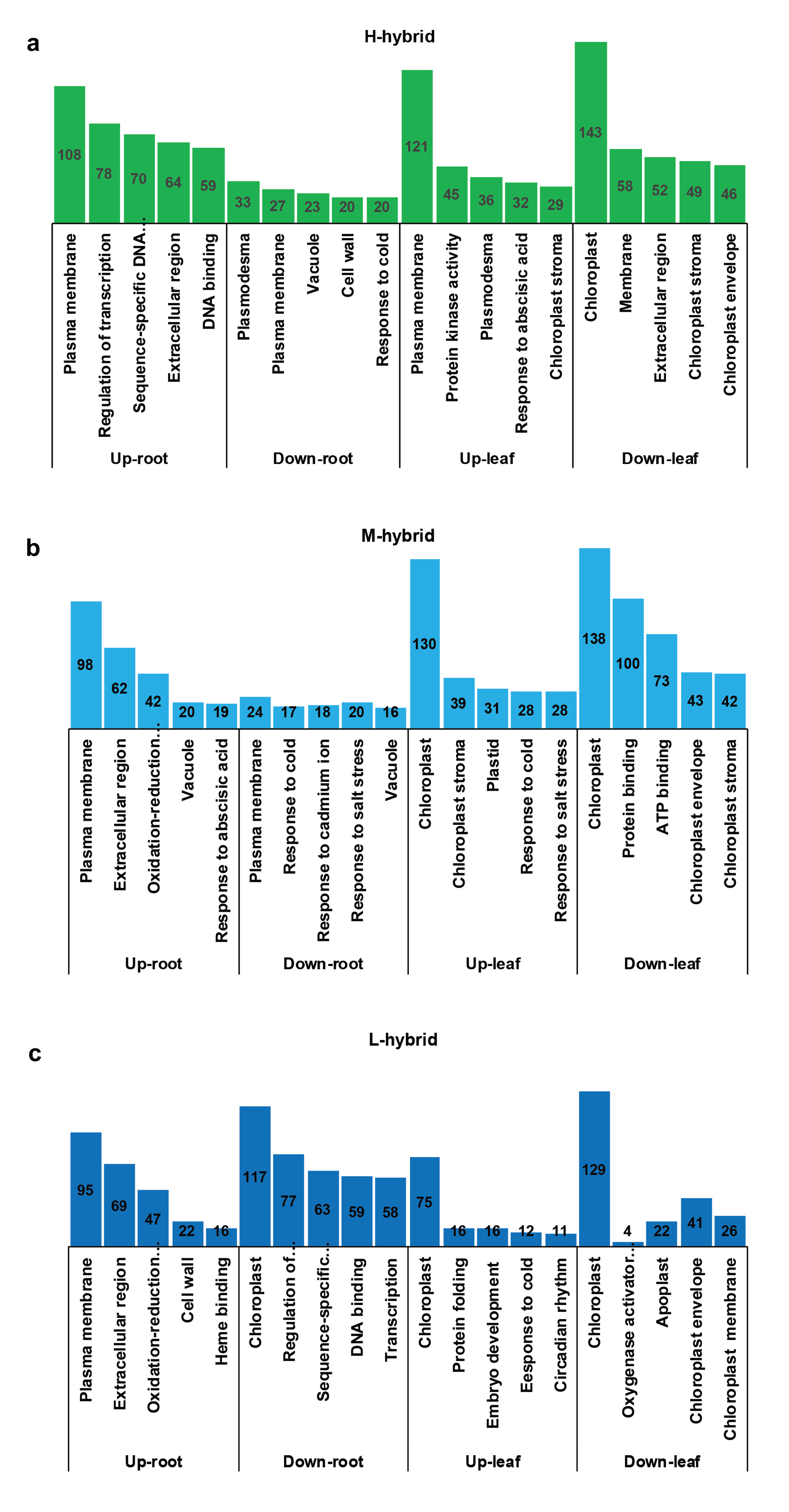
Functional annotations of overdominant genes
To understand the functions of genes with overdominant expressions in biomass heterosis, GO and KEGG enrichment analysis was implemented in root and leaf of hybrids. GO enrichment analysis (p-value < 0.01) of overdominant genes in root of high hybrid (H) relative to its parents revealed most of the upregulated genes were involved in functions related to plasma membrane, regulation of transcription/DNA-template, and extracellular region. Conversely, downregulated genes were enriched in plasmodesma, integral component of plasma membrane, and vacuole (FigS3a: Additional file 11). KEGG pathway enrichment analysis (p-value < 0.05) of overdominant genes showed most of the upregulated genes were enriched in porphyrin & chlorophyll metabolism, phenylpropanoid biosynthesis, and oxidative phosphorylation pathways (Fig. 4a: Additional file 12). But, the majority of downregulated genes were enriched in starch & sucrose metabolism, phenylpropanoid biosynthesis, and circadian rhythm plant (Fig. 4b: Additional file 12). In the leaf of H hybrid, most of the overdominant upregulated genes were involved in functions of plasma membrane, protein serine/threonine kinase activity, and plasmodesma, while downregulated genes were involved in cellular functions of chloroplast, membrane, and extracellular region (FigS3a: Additional file 13). Enriched pathways of overdominant genes found upregulated genes were enriched in plant-pathogen interaction, plant hormone signal transduction, and circadian rhythm plant (Fig. 4a: Additional file 14). In contrast, significant pathways for downregulated genes were plant hormone signal transduction, carbon metabolism, and circadian rhythm plant (Fig 4b: Additional file 14).
The results of GO enrichment analysis of overdominant genes in the root of medium hybrid (M) compared with its parents uncovered that upregulated genes were enriched in plasma membrane, extracellular region and oxidation-reduction process. In contrast, downregulated genes were involved in following functions e.g. integral component of the plasma membrane and response to salt stress (FigS3b: Additional file 15). Pathway enrichment analysis of overdominant genes found upregulated genes were enriched in phenylpropanoid biosynthesis, amino & nucleotide sugar metabolism, and porphyrin & chlorophyll metabolism (Fig 4a: Additional file 16). For downregulated genes, enriched pathways were circadian rhythm plant, glycosaminoglycan degradation, and ascorbate and aldarate metabolism (Fig. 4b: Additional file 16). GO enrichment analysis of overdominant genes in leaf of M hybrid revealed most of upregulated genes performed functions related to chloroplast, chloroplast stroma, and plastid. However, downregulated genes involved in the following functions such as chloroplast, protein binding, and ATP binding (FigS3b: Additional file 17). Pathway enrichment analysis of overdominant genes revealed most of upregulated genes were found in carbon metabolism, phagosome, and circadian rhythm plant (Fig. 4a: Additional file 18). On the other hand, downregulated genes were enriched in planted hormone signal transduction, endocytosis, and carbon metabolism (Fig. 4b: Additional file 18).
GO enrichment analysis of overdominant genes in the root of low hybrid (L) compared with its parents found following enriched terms e.g. plasma membrane, extracellular region, and oxidation-reduction process. The functions of downregulated genes were enriched for chloroplast, regulation of transcription/DNA-template, and transcription factor activity/sequence-specific DNA binding (FigS3c: Additional file 19). Enriched pathways of overdominant upregulated genes were phenylpropanoid biosynthesis, porphyrin & chlorophyll metabolism, and pentose & glucuronate interconversions (Fig. 4a: Additional file 20). On the other hand, most of downregulated genes were enriched in circadian rhythm plant, carbon metabolism, and starch & sucrose metabolism (Fig. 4b). In the leaf of L hybrid, GO enrichment analysis of overdominant genes showed functions of upregulated genes were enriched in chloroplast, protein folding, and embryo development ending in seed. In contrast, majority of the downregulated genes were involved in cellular function of chloroplast, membrane, and chloroplast stroma (FigS3c: Additional file 21). The pathway enrichment analysis of overdominant genes uncovered majority of upregulated genes were involved in protein processing in the endoplasmic reticulum, spliceosome, and circadian rhythm plant (Fig. 4a: Additional file 22). For downregulated genes, enriched pathways were carbon metabolism, plant hormone signal transduction, and circadian rhythm plant (Fig. 4b: Additional file 22).
The comparison of overdominance genes between contrasting hybrids
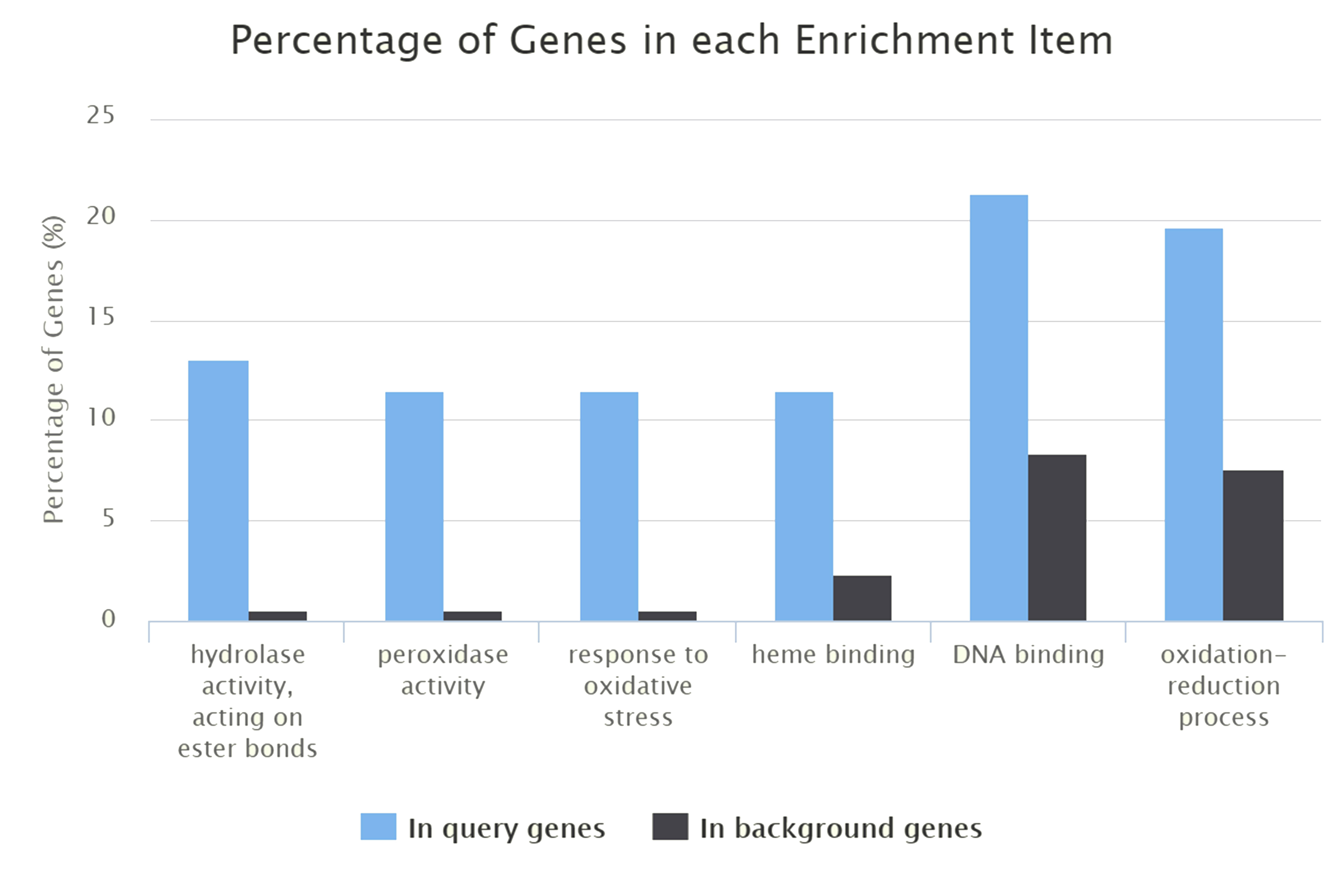
The results of KEGG revealed that most of the overdominant genes of high, medium and low hybrids relative to their parents were enriched in similar pathways. We performed comparison analysis between high, medium and low hybrids to see unique and common overdominant expressed genes in the above mentioned pathways. The results showed that 44 genes in leaf and 29 genes in root were common in comparison of all hybrids (Fig. S4: Additional file 23). On the other hand, many genes were also overlapped between comparisons of two hybrids. The reason behind these results might be the common female parent in hybrids. The expression level of genes that showed overdominant expression in all hybrids relative to their inbred parents is presented in Fig. 5. We found most of root genes were belonging to circadian rhythm, phenylpropanoid biosynthesis, and porphyrin & chlorophyll metabolism, while leaf genes were enriched in circadian rhythm, plant hormone signal transduction, and carbon metabolism. Majority of these genes were involved in functions related to DNA binding, oxidation-reduction process, heme binding, and response to oxidative stress (Fig. S5). We presumed phenomenal changes in these pathways associated genes may be transformed biomass vigor in hybrids of cotton.
The circadian rhythm, metabolic processes, and biomass vigor
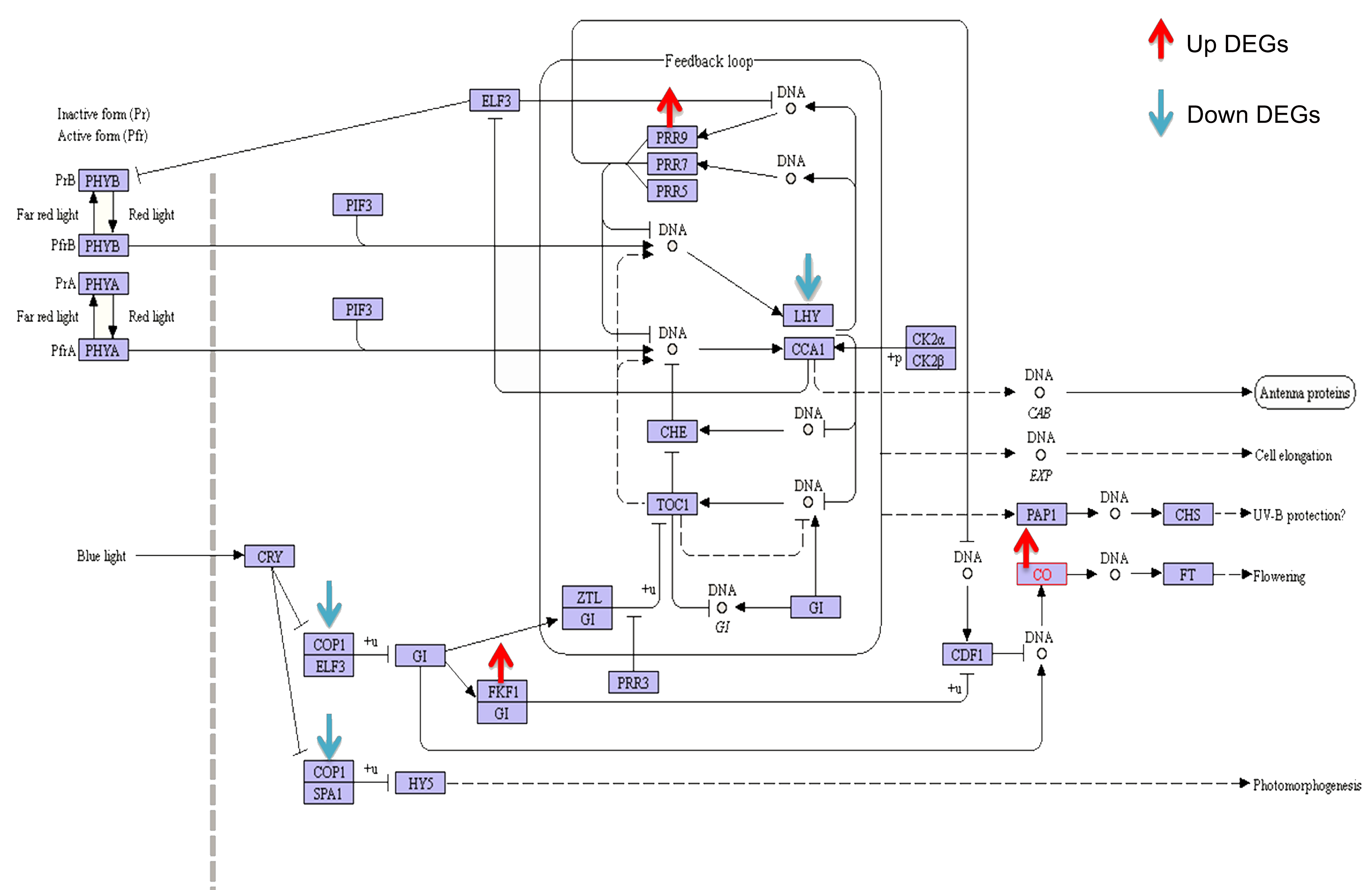
Through compression analysis between hybrids, we found circadian rhythm plant pathway contained many overdominant genes in root and leaf of all hybrids. Genes in this pathway had tissue-specific expression except of three (Gh_A12G1061, Gh_A12G1062, and Gh_D12G1185). In this pathway, six genes (Gh_A11G0926, Gh_A12G1061, Gh_A12G1062, Gh_D12G1185, Gh_D12G1184, and Gh_D11G1068) related to MYB domain transcription factor, encoding LHY protein showed downregulation in root of hybrids (Fig. S6). In contrast, five genes (Gh_A12G1061, Gh_A12G1062, Gh_D12G1185, Gh_A09G1504, and Gh_D09G1515) encoding LHY protein showed downregulated expression in leaf of hybrids. The results indicated four genes (Gh_A08G0451, Gh_D01G0200, Gh_D07G0867, and Gh_D11G1518) associated with transcript factor CO-like showed upregulation in leaf of hybrids (Fig. S6). Genes (Gh_A05G0944 and Gh_D05G1029) named as CIA2 (CHLOROPLAST IMPORT APPARATUS 2) associated with PSEUDO-RESPONSE REGULATOR9 (PRR9) also showed similar results in leaf of hybrids (Fig. S5). In addition to circadian rhythm, several genes (Gh_A03G0944, Gh_A09G1415, Gh_A11G1859, Gh_A12G1915, and Gh_D02G1327) in phenylpropanoid biosynthesis related to peroxidase superfamily protein, and many genes from porphyrin &chlorophyll metabolism showed upregulated expressions in root of hybrids. Furthermore, many genes of plant hormone signal transduction pathways linked to AUX/IAA transcriptional regulator family protein and genes from carbon metabolism pathway also found with downregulated expressions in leaf of hybrids. To be concise, all these results suggest genes in circadian rhythm together with other metabolic process performed overdominance that might change root and leaf growth, and ultimately lead to altered biomass vigor in hybrid cotton.
qRT-PCR analysis
Twelve overdominant genes from circadian rhythm plant were selected to validate RNA-seq data by real-time qRT- PCR analysis. All selected genes had overdominance expression in high, medium, and low hybrids relative to their parents in RNA-seq results. We selected five genes (Gh_A12G1061, Gh_A12G1062, Gh_D12G1184, Gh_D11G1068, and Gh_D13G1939) from root and one gene (Gh_A09G1504) from leaf with downregulated expressions in hybrids, while six genes (Gh_A08G0451, Gh_D01G0200,Gh_D11G1518,Gh_D05G1029 Gh_D07G0867,Gh_A05G0944, and Gh_D12G1525) were selected from leaf with upregulated expressions in hybrids. It was found that the expression trends as calculated by qRT-PCR were consistent with RNA-seq data, thus confirming the accuracy of the RNA-seq results in this study (Fig. 6).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}