Distinct phenotypes of autotetraploid cassava compared with its diploid progenitor
Autopolyploid cassava (4x) had previously been generated by colchicine-induced chromosome doubling from the diploid (2x) cultivar ‘Xinxuan 048’ [47]. The ploidy levels of the generated 4x plants were detected by the flow cytometry analysis, and then chromosome counting in root-tip cells confirmed the results of flow cytometry analysis. The 2x cassava had 36 chromosomes and the 4x had 72 chromosomes [47]. The plants were propagated and maintained for two years. The leaves of the 4x cassava were significantly larger, greater and darker green than those of 2x cassava (Additional file 1: Fig. S1) and showed improved drought resistance [23].
Single base-resolution maps of DNA methylation for diploid and autotetraploid cassava
In order to investigate the potential role of DNA methylation in response to autotetraploidy, the methylomes of 2x and 4x cassava leaf were decoded and analyzed. To assess variability, three biological replicates were generated of 2x and 4x, respectively. Genome mapping analysis showed that 113,078,644 (73.50%), 109,732,951 (73.44%), and 111,434,206 (74.25%) clean reads of each three replicate of 2x sample, while 119,723,975 (74.70%), 96,864,854 (75.06%) and 110,185,024 (75.65%) clean reads could be mapped for each replicate library. The sequence depth of all the samples were more than 24×. More than 99% of cytosines were altered, which indicated that a high rate of conversion (Additional file 2: Table S1). In addition, pearson correlation coefficients between the three replicates of 2x and 4x were found to be between 0.96 and 0.97 (Additional file 3: Fig. S2). All the data indicated that the quality of sequencing was satisfactory with subsequent analysis.
Among all the sequenced C sites, the 2x and 4x genome presented 67.09% and 66.94% (mCGs), 49.03% and 48.67% (mCHG), and 5.88% and 5.74% (mCHH) in three sequences contexts, respectively (Additional file 4: Fig. S3), which reflected all three contexts methylation did not show any significant variation after WGD (P > 0.05). At the chromosome-scale scale, it was discovered that the methylation levels of three contexts were all predominantly highly pericentromeric heterochromatin regions and methylation levels of all three contexts in 2x and 4x cassava were similar to one another (Additional file 5: Fig. S4).
Landscape of PCGs, lncRNAs and TEs methylation
To characterize the DNA methylation patterns in different cassava genomic regions, we constructed the methylation profiles within PCGs, lncRNAs and TEs, together with 2-kb regions flanking the genes, using the same cut-off lengths (Fig. 1). For PCGs, lncRNAs and TE regions, relatively high methylation levels were identified in CG context, followed by CHG and CHH contexts. Methylation patterns across PCGs (Fig. 1a) and TEs (Fig. 1c) in our study was consistent with that of Wang et al. [48]. The average methylation levels of TEs were much higher than that of PCGs and lncRNAs, indicating TEs were easier to be methylated. There were no differences of CG methylation levels in PCGs bodies in 2x and 4x cassava (Wilcoxon rank sum test, P value = 0.7465, n = 33,033) (Fig. 1a). For mCHG and mCHH sites, hypermethylation state of PCGs bodies was observed in 2x cassava (Wilcoxon rank sum test, mCHG P value = 0.00010365, mCHH P value = 9.76E-45, n = 33,033) (Fig. 1a). The methylation state of lncRNA in all three contexts were higher than those of PCGs (Fig. 1b). Similarity, 4x cassava had decreased CG, CHG and CHH methylation levels relative to 2x across lncRNA body regions and flanking regions (Wilcoxon rank sum test, mCG P value = 0.0269798, mCHG P value = 0.73398116, mCHH P value = 0.0118822, n = 13,533) (Fig. 1b). We also found that TEs had decreased methylation levels in CHG and CHH contexts in 4x cassava compared to 2x (Wilcoxon rank sum test, mCG P value = 8.98E-06, mCHG P value = 4.23E-21, mCHH P value = 5.13E-75, n = 617,861) (Fig. 1c). In summary, WGD may have widespread influence on methylation levels of both PCGs and lncRNAs body regions in CHG and CHH contexts, rather than CG context.
Consequently, we detected the methylation differences of the two classes of TEs. All the 13 orders of TEs had unique average methylation distribution, and exhibited hypermethylation state in body regions than flanking regions in all three contexts. In light of the proportion of SINE, Stowaway, Harbinger and other_DNA is too small in the cassava genome, so the four types of TEs were not be considered for further analysis (Additional file 6: Table S2). Obviously, all the class I TEs including Copia, Gypsy, LINE, and other_LTR exhibited hypermethylation levels in mCHG and mCHH sites in 2x cassava (Fig. 1d, e). We also found that almost all the body regions of class II TEs from 4x cassava had increased CG methylation levels except those of En_Spm and MuLE_MuDR. Moreover, body regions of MITE from 4x cassava were hypermethylated in all three contexts. Although En_Spm, Helitron, hAT and MULE-MuDR exhibited hypermethylation levels in mCHG and mCHH sites in 2x cassava, methylation levels in CHG and CHH contexts were much lower than those in CG context (Fig. 1f, g).
Unlike class I TEs, class II TEs tended to localize in euchromatin regions where PCGs were actively expressed. Genome-wide changes of TEs methylation levels may affect expression of neighboring PCGs that were inserted and surrounded by class II TEs after WGD. On the other hand, the proportion of TE-overlapped lncRNAs to all lncRNAs detected is 40% (Additional file 7: Fig. S5), which supported a major of lncRNAs are either derived from TEs or contain TEs remnants [38-40]. Genome-wide alteration of class I TEs methylation levels may affect expression of adjacent lncRNAs as a result of autotetraploidization. Therefore, it was sensible and necessary to combine TE methylation and PCG and lncRNA expression to examine the epigenetic responses to WGD.
Gene methylation is associated with gene activity
In view of the difference of methylation between 2x and 4x cassava after genome doubling, we attempted to understand whether PCG- and lncRNA-expression levels were influenced by DNA methylation. A total of 33,030 PCGs and 13,517 lncRNAs assembled from the lncRNA-seq data in ‘Xinxuan 048’ were divided into four quartiles from high-expressed group, low-expressed group, middle-expressed group and none-expressed group based on their expression levels according to the criteria of Yan et al. [49]. PCGs with relative high CG body methylation level showed high expression, but relatively low expression in the flanking regions in 2x and 4x cassava (Fig. 2a). In contrast, PCGs with high-expressed showed the lowest CHG and CHH methylation levels, middle-expressed PCGs displayed the second highest CHG methylation levels, and PCGs with non-expressed displayed the highest methylation level in the two cassava genotypes (Fig. 2b, c). The results indicated that positive correlation was observed between DNA CG methylation and PCG-expression levels, there was negative correlation for CHG and CHH methylation levels and PCG expression in 2x and 4x cassava.
As is shown in Fig. 2d, there was irregular of DNA methylation of mCG against global lncRNA expression because the four expressional graph against the mCG site of lncRNAs twined together seriously in 2x and 4x cassava. LncRNAs with high-expressed showed the highest CHG methylation levels, middle-expressed lncRNAs displayed the second highest CHG methylation levels, and lncRNAs with non-expressed displayed almost the same relative low methylation level as that of the low-expressed lncRNAs. CHG methylation throughout lncRNA body regions together with flanking regions is positive with lncRNA expression. mCHH sites of upstream and downstream regions is positive correlated with lncRNA expression, lncRNAs with middle-expressed throughout lncRNA body region showed the highest methylation levels in CHH context in cassava. Both of low-expressed and none-expressed lncRNA displayed the same CHG and CHH methylation levels, which were lower than those of lncRNAs with high-expressed and middle-expressed (Fig. 2e, f). The results indicated that no correlation was observed between DNA CG methylation and lncRNA-expression levels, there was positive correlation for mCHG and mCHH methylation levels and lncRNA expression in 2x and 4x cassava.
Transposons with changed DNA methylation caused by WGD altered the expression of nearby PCGs and lncRNAs
The results of the transcriptome analysis indicated that 33,030 PCGs and 13,517 lncRNAs were detected in this study, which formed the dataset for the subsequent study. Comparison of expression level of PCGs and lncRNAs between 2x and 4x cassava indicated that only 359 PCGs and 402 lncRNAs were differentially expressed, respectively. That is, compared with 2x cassava, a large number of PCGs and lncRNAs were not significantly expressed in 4x cassava genotype that has obtained double alleles (Wilcoxon rank sum test, PCG P value = 0.5061817, n = 33,030; lncRNA P value < 0.003689, n = 13,517). Compared with 2x cassava, there were 173 PCGs up-regulated, and there were 186 PCGs down-regulated (Additional file 8: Fig. S6a); there were 204 lncRNAs up-regulated, and 198 lncRNAs down-regulated in 4x cassava (Additional file 8: Fig. S6b).
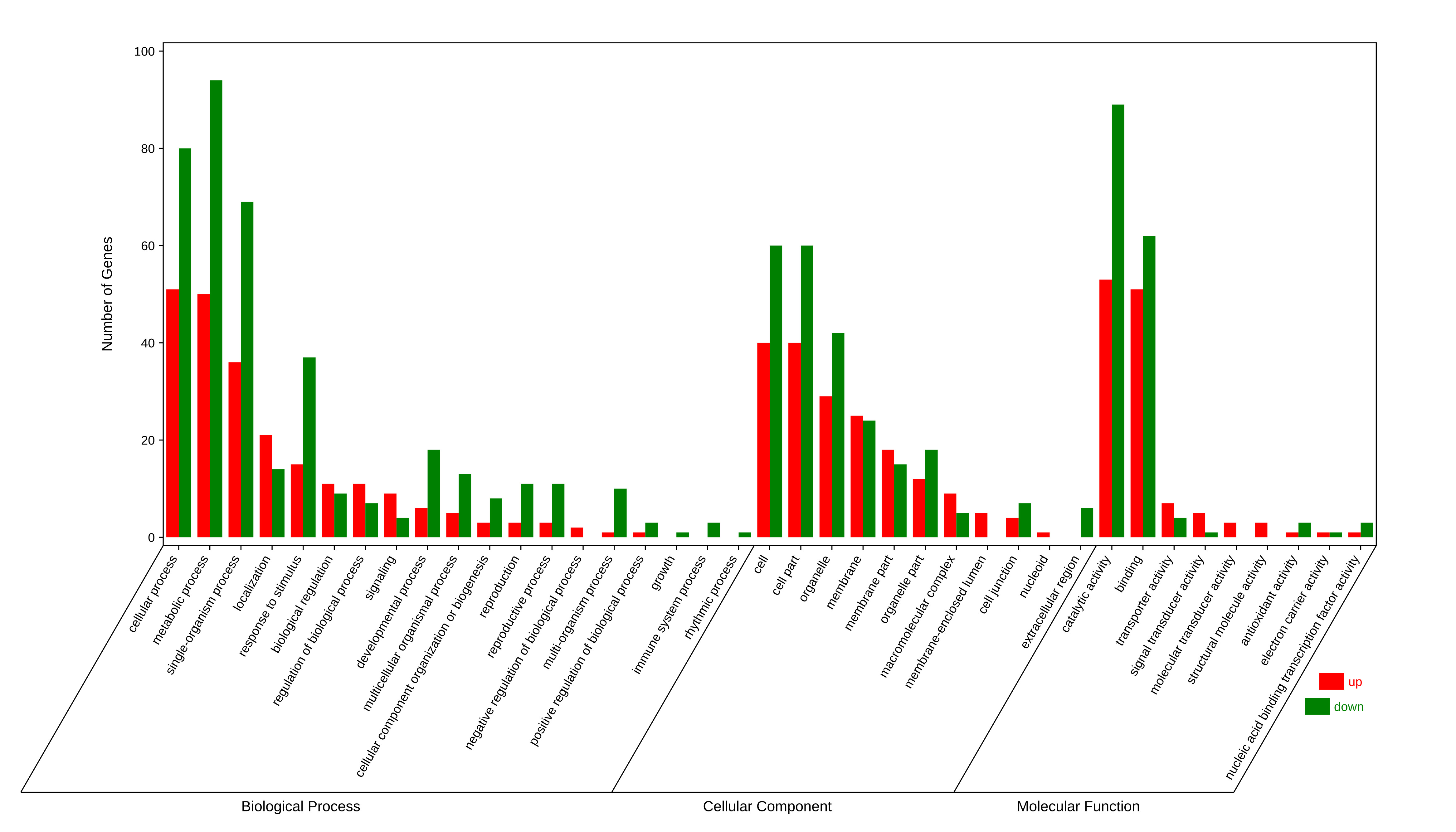
The differentially expressed PCGs were then used to be GO enrichment analysis. The 173 up-regulated PCGs in 4x cassava were involved in ‘cellular process’, ‘metabolic process’, ‘response to stimulus’, ‘reproduction’, etc. (Additional file 9: Fig. S7). The 186 down-regulated PCGs fell into ‘metabolic process’, ‘cellular process’, ‘response to stimulus’, ‘development process’, etc. (Additional file 9: Fig. S7).
To identify the two hypotheses mentioned above, the question is whether TEs may affect expression of neighboring PCGs and lncRNAs that were involved in WGD-induced variation in cassava. To address this question, 33,030 PCGs and 13,517 lncRNAs were used to examine whether they were inserted or surrounded by TEs. The results showed that approximately 48% of PCGs had TEs insertion into their bodies. Most of the TE insertions were DNA transposons, about 28% and 62% of PCGs were inserted by DNA transposons within bodies and 8-kb flanking regions, respectively (Fig. 3a). About 40% of lncRNAs had TEs into their bodies, 80% of lncRNAs were inserted by TEs within 2 kb, 94% lncRNAs overlapped with TEs within 4-kb flanking regions. Most of the TEs insertion into lncRNAs were retrotransposons, 25% and 52% of lncRNAs had retrotransposons insertion into their bodies and within 8-kb flanking regions, respectively (Fig. 3b). Moreover, we found that in 2x and 4x cassava, PCGs without neighboring TEs were expressed at higher levels than those inserted or surrounded by TEs (Fig. 3c). The expression levels of lncRNAs inserted or surrounded by TEs were higher than those without nearby TEs in the two cassava genotypes (Fig. 3d). The average PCG-expression level was positively correlated with the distance to the closest TE (2x P value = 4.36E-148; 4x P value = 4.69E-156) (Fig. 3e), for both 2x and 4x cassava, in contrast, the average lncRNA-expression level was negatively correlated with the distance to the closet TE (2x P value = 4.41E-25; 4x P value = 1.80E-39) (Fig. 3f). In addition, the average PCG-expression level was negatively correlated with the number of TEs within 4-kb flanking regions (2x P value = 6.38E-32; 4x P value = 5.23E-33) (Fig. 3g), however lncRNA-expression level was positively correlated with the number of TEs within 4-kb flanking regions in the two genotype cassavas (2x P value = 0.002433; 4x P value = 0.0007531) (Fig. 3h). Collectively, these results indicated that PCG-expression levels were suppressed by the abundance and physical distances from adjacent TEs, and lncRNA-expression levels were activated by the abundance and physical distances from proximal TEs.
Considering expression level of adjacent PCGs were negatively correlated with the state of methylated TEs in Arabidopsis thaliana and rice [21, 50], we compared DNA methylation of TEs from the whole genome, gene body, and flanking 4-kb regions between 2x and 4x cassava. Methylation of class II TEs, for both PCGs and lncRNAs, were lower than that of class I TEs for all three contexts (Fig. 4a, b), and methylation levels of TEs or class II TEs for PCGs in 4x cassava were higher than those of 2x except for flanking TEs in CHH context (Fig. 4a). Nevertheless, TEs or class I TEs inserted or surrounding lncRNAs from 2x cassava exhibited hypermethylation in CHG and CHH contexts (Fig. 4b).
We divided PCG-flanking regions into different bins and compared methylation levels between two TE classes located within them (Fig. 4c). Obviously, methylation levels of class II TEs nearby PCGs were lower than that of class I TEs in all three contexts. The CG methylation levels of class II TEs gradually decreased with increased distances from PCGs, and the valley of CG methylation levels of TEs appeared within 0.5-kb flanking regions. For mCHG and mCHH sites, class II TEs methylation levels gradually increased with increased distances from PCGs. Notably, 4x cassava exhibited hypermethylation in CG and CHH contexts in PCG-flanking regions of class II TEs. To sum up, hypomethylation of class II TEs inserted in PCGs body regions in CHG and CHH contexts and hypermethylation of class II TEs neighboring PCGs in CG and mCHH contexts, may be a direct response factor to overcome genome-dosage effects following WGD in 4x cassava.
Parallelly, we divided lncRNA-flanking regions into different bins and compared methylation levels between two TE classes located within them (Fig. 4d). Similarity with that of PCGs, methylation levels of class II TEs nearby lncRNA in all three contexts were lower than those of class I TEs. The profile of methylation levels of class I TEs did not show always rising for lncRNAs, which was different from that of PCGs. The CHG methylation levels of class I TEs had the same dynamic change with that of CHH methylation levels with increased distances from lncRNAs excluding within 0 to 0.5-kb flanking regions. The CG methylation levels of class I TEs appeared to be higher in 4x than that of 2x in 0~2.5-kb regions, however, CG methylation levels of class I TEs were lower in 4x in the flanking regions after 0~2.5-kb regions. Critically, in 4x cassava, class I TEs exhibited hypomethylation state in the CHG and CHH contexts compared with 2x in lncRNA-flanking regions, with the exception that CHG methylation state in ~1.5 to 3.0-kb lncRNA-flanking regions observed in 4x was the same as that of 2x, and this result was consistent with the genome-wide methylation level of the five types of class I TEs (Fig. 1d, e). Taking together, reduction of CHG and CHH methylation levels of class I TEs nearby lncRNAs in autotetraploid cassava may be a mechanism that suppressed expression of adjacent lncRNAs in autotetraploid cassava with double alleles from the diploid line responding to genome-wide lncRNA dosage effects after WGD.
Usually, retrotransposons of the Gypsy and Copia superfamilies are the major partitions of lncRNA in plant genome, especially for lincRNA [39, 40, 51]. Then, we calculated the percentage of the diversity classes of lncRNAs detected in this study, and found that lincRNAs (or called intergenic lncRNA) accounted for the largest proportion (42%) of the whole lncRNAs (Additional File 10: Fig. S8). We also found that the lncRNA loci contained more Gypsy and Copia segments than the other TEs at the exon and intron sequences together with 8-kb flanking regions in the cassava genome. Gypsy showed the largest proportion of lncRNA-overlapped TEs, followed by Copia, due to its largest share of TEs in the cassava genome (Additional File 11: Fig. S9). In order to understand whether the hypomethylated state of Gypsy or Copia was the effector overcoming genome-dosage effects in autotetraploid cassava, we depicted the correlation diagram of the DNA methylation level of Gypsy and Copia related to the distance from the closest lncRNA and lincRNA in 2x and 4x cassava, respectively (Fig. 4e-h). The results indicated that only the profile of methylation levels of Gypsy segment related to the closest lincRNA was found to be almost the same with that of class I TEs related to the closet lncRNA in all three contexts, with the exception that CHG methylation in 1.5~3.0-kb lncRNA-flanking regions observed in 4x was slightly higher than that of 2x, after all, CHG methylation levels of class I TEs in 4x cassava were the same as that of 2x in this region (Fig. 4e).
Comparisons of DMRs between 2x and 4x cassava
To investigate the potential effect of WGD, we identified a total number of 922 CG, 608 CHG and 51 CHH DMRs (Fig. 5a). 64.92% of mCG sites were hypermethylated while 43.09% of mCHG and 17.05% of mCHH sites exhibited hypomethylation in 4x cassava (Fig. 5a).
At the whole-genome level, most DMRs came from the mCG context, hardly any CHH hyper-DMRs existed in 4x cassava (Fig. 5b). The number of CG hyper-DMRs in 2x cassava was half that in 4x cassava, while the number of CHG-DMRs and CHH-DMRs in 2x is more than that in 4x (Fig. 5b). Genome-wide analysis showed that DMR of all three contexts were inclined to localize in intergenic and TEs regions rather than PCGs and lncRNAs, CHG-DMR within lncRNAs of 4x cassava increased in comparison with 2x cassava, there is no CHH hyper-DMR lncRNAs in 4x cassava (Fig. 5c). Analysis of PCG-flanking and lncRNA-flanking 4-kb regions revealed that the distribution of DMRs was similar to that throughout the whole genome (Fig. 5c).
{kind=link}
{kind=link}