2.1) Experimental Design
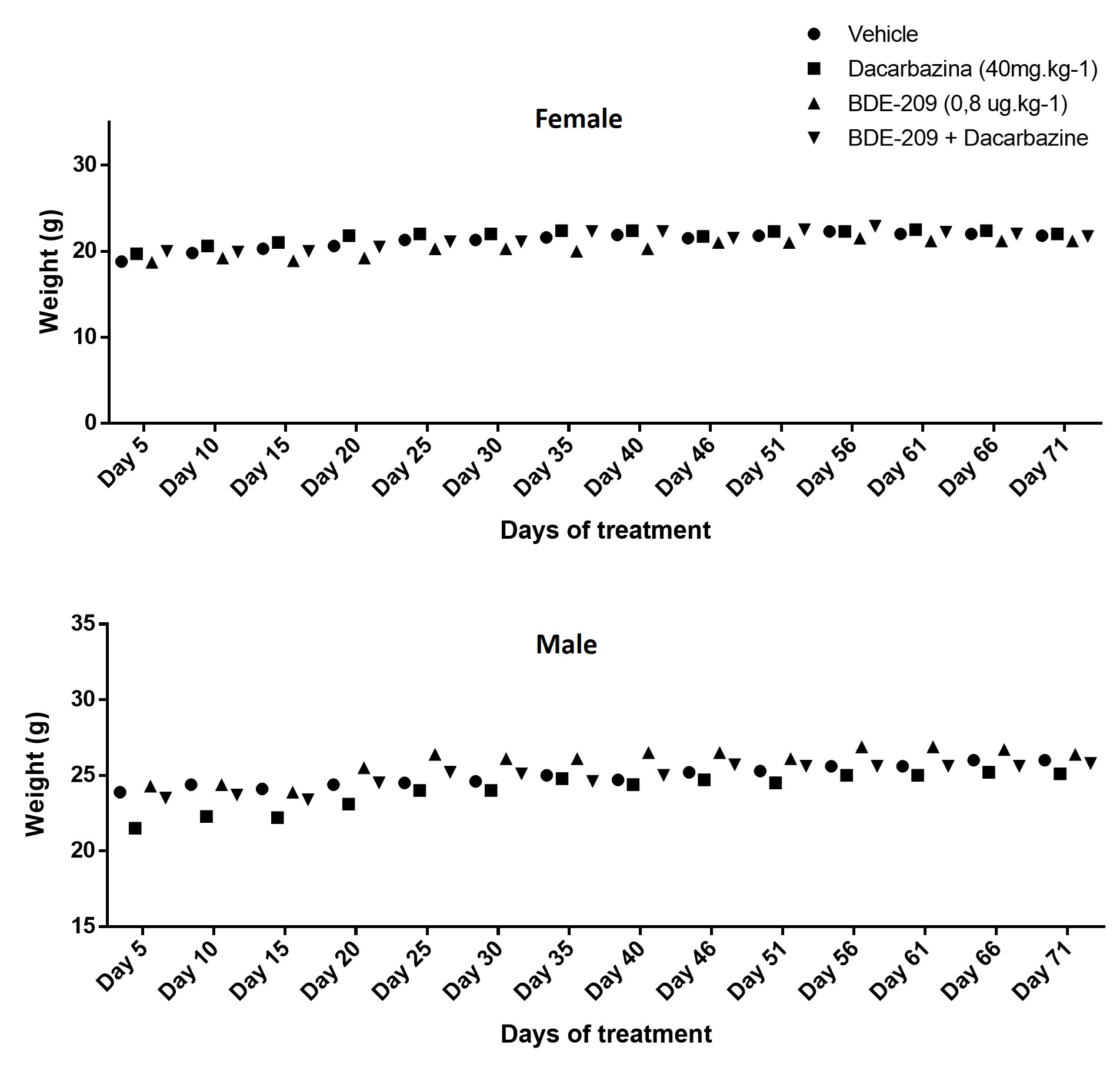
The current experimental design was approved by the Ethics Committee on Animal Experimentation at Federal University of Parana (certificate no. #1040/2017). Seven-week-old C57BL/6 mice (50 males and 50 females, no ovariectomized or orchiectomized) were maintained in cages (5 animals per cage). Mice of the same sex were randomly distributed in 3 groups: animals maintained without exposure (no gavage was performed); animals exposed only to corn oil by gavage (vehicle); and animals exposed to BDE-209 (0.8 µg.kg− 1 in corn oil by gavage every 5 days; Fig. 1). After 45 days (nine doses), the animas were injected with B16-F10 cells (2.5×105 cells in 100 µL of PBS) via caudal vein. In a second phase, five groups (10 animals per group) were organized as follow: control (without gavage), vehicle (gavage with corn oil), dacarbazine (only treated with dacarbazine), BDE-209 (only gavage with BDE-209) and BDE-209 + dacarbazine (gavage with BDE-209 and treatment with dacarbazine; Fig. 1). The chemotherapy treatment was performed with 40 mg/kg of dacarbazine every 3 days (six doses by intraperitoneal injections) after the 49th day of experiment. Finally, on the 66th day, the mice were anesthetized and euthanized (10 mg.kg− 1 of xylazine, 100 mg.kg− 1 of ketamine), and the blood, lung, liver, kidney and brain were weighed and sampled for the analyses (Fig. 1).
B16-F10 mouse melanoma cells (American Type Culture Collection - Manassas, USA) were cultured in DMEM culture medium (Lonza Bio Whittaker - Basel, Switzerland), supplement with 0.1 M of HEPES (Lonza Bio Whittaker) and gentamicin (40 mg/L) and fetal bovine serum (10%). Mycoplasma contamination was routinely tested.
2.2) Blood Analysis
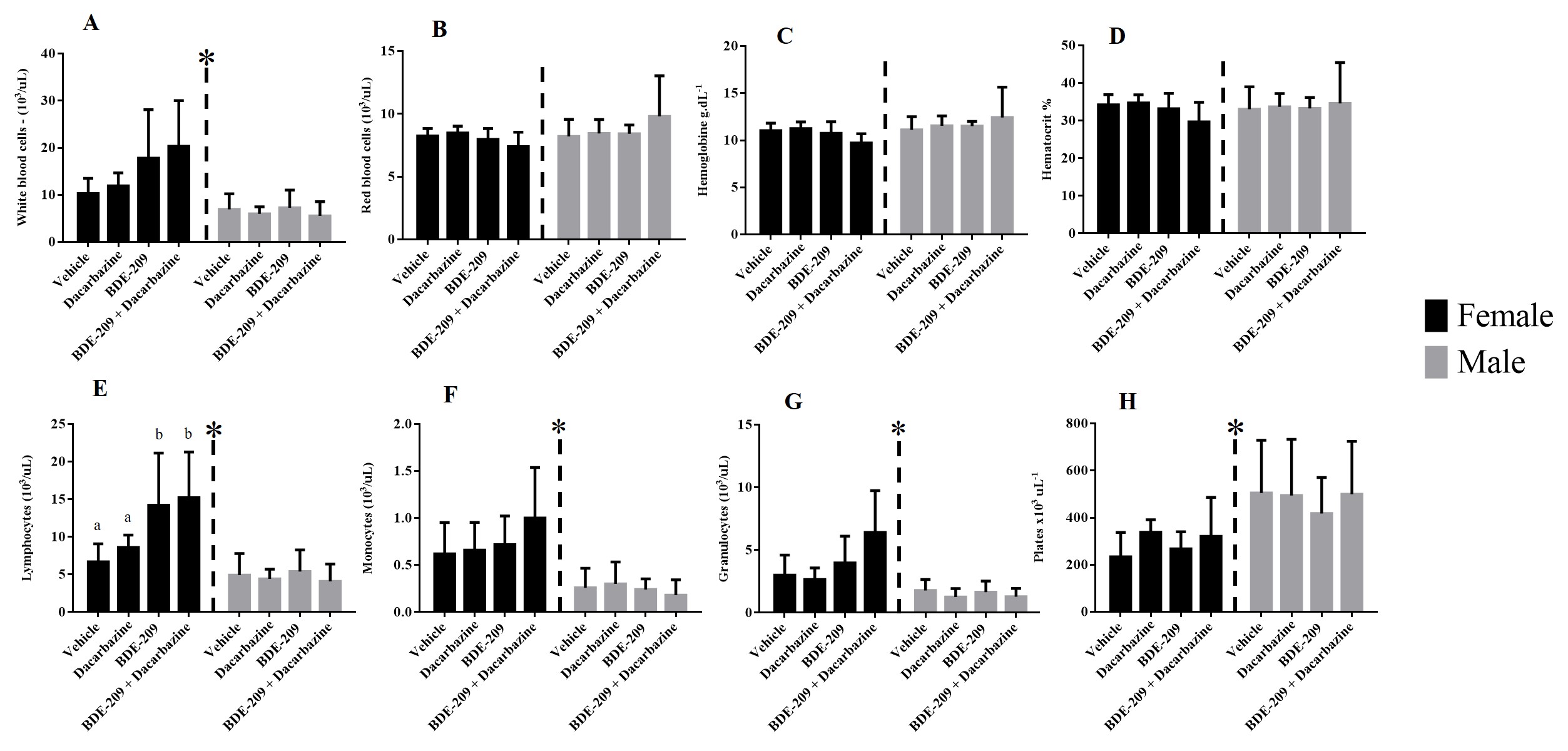
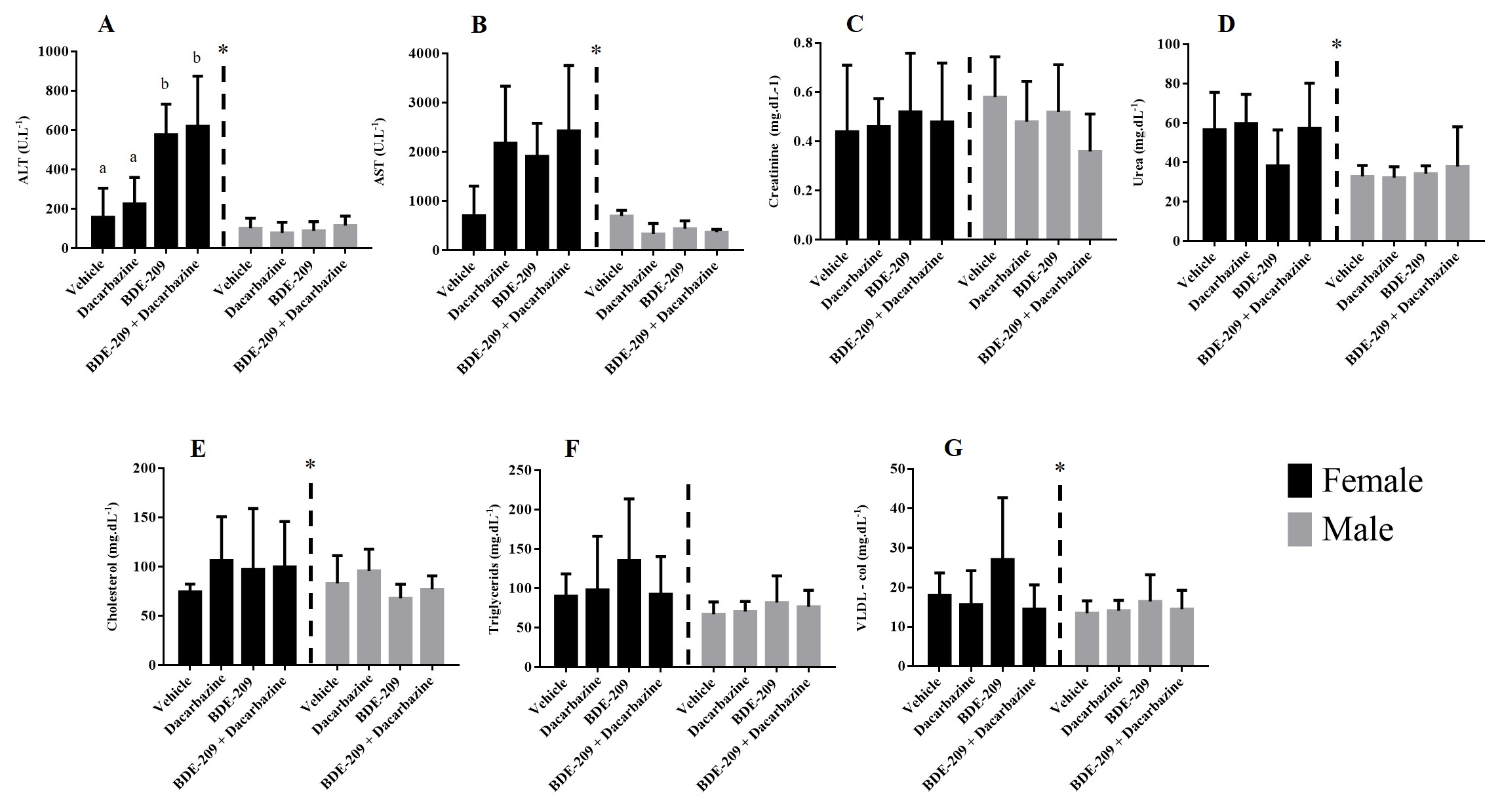
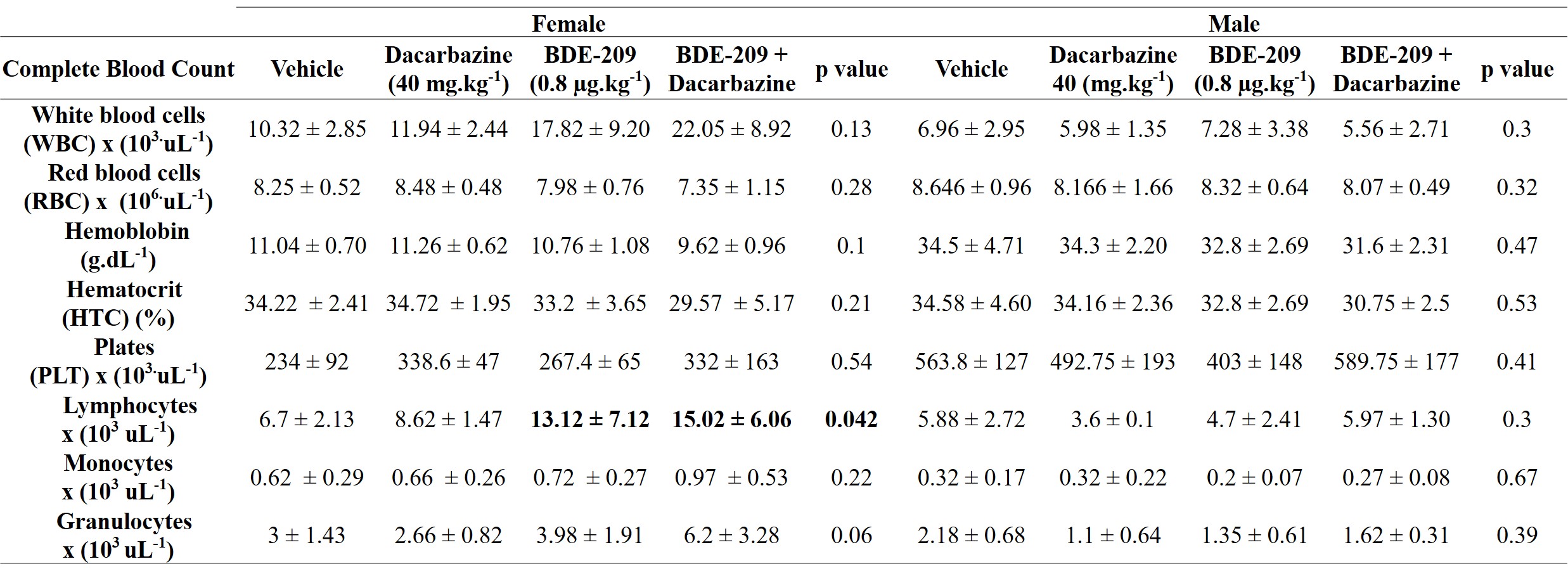
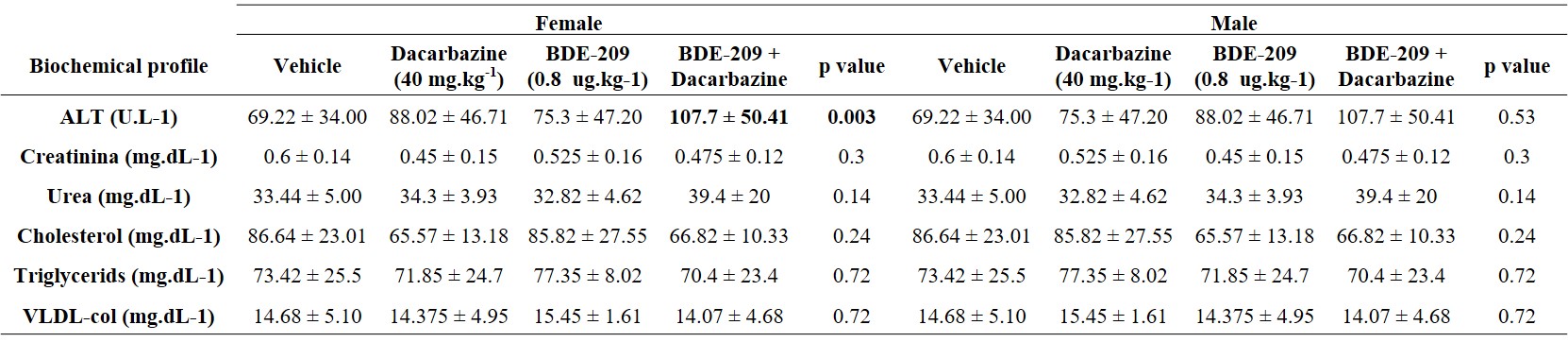
The blood was sampled via intracardiac route and a complete blood cell counting (CBC) was performed (total leukocytes, red blood cells, hemoglobin, hematocrit, platelets, segmented, stems, eosinophil, and lymphocytes) at the Veterinary Clinical Pathology Laboratory at Federal University of Parana, Brazil. The plasma was centrifuged (3,000 g for 10 min) and utilized for biochemical profile determination (alanine aminotransferase-ALT, aspartate aminotransferase-AST, alkaline phosphatase, cholesterol, triglycerides, creatinine, and urea) using a Mindray BS-200 Automated Benchtop Biochemistry Analyzer.
2.3) Biochemical biomarkers analysis
The lung, liver and kidney were sampled and stored at -80°C. Then, 0.2 g of each organ were homogenized in 2 mL of Tris-buffer (20 mM HCl, 0.1 M EDTA, 1 mM PMSF, pH 7.4) and centrifuged at 12,000 g (4ºC) for 30 min. The supernatant was recovered, and the total protein concentration was determined according to Bradford (1976).
Non-protein thiols (NPT) concentration was measured in supernatants after protein precipitation with 10% trichloroacetic acid and centrifugation (1,000 g for 15 min and 4°C). Supernatant (50 µL) and 230 µL of Tris (0.4 M, pH 8.9) were placed in a 96-well microplate, followed by addition of 20 µL of 2.5 mM DTNB [5, 5'-dithiobis (2-nitrobenzoic acid) in 25% methanol. Absorbances were determined at 415 nm and NPT concentration was calculated by comparison with the standard curve for GSH (Sedlak and Lindsay, 1968).
Superoxide Dismutase (SOD) activity was measured in supernatants (1.0 mg protein/mL) after addition of ethanol (1:5) and new centrifugation (9,000 g, 4ºC for 30 min). Supernatant (20 µL) and 35 µL of 572 µM NBT chloride in 0.1 mM EDTA were mixed in a 96-well microplate. The reaction was initiated through the addition of 145 µL of 51 mM hydroxylamine chloride in 0.5 M sodium carbonate (pH 10.2). The reduction of NBT by O2− to blue formazan was measured spectrophotometrically as a constant increase of absorbance at 560 nm for 30 min. The rate of NBT reduction in the absence of samples was used as the reference rate. One unit of SOD was defined as the enzymatic activity able to inhibit the reduction of NBT to 50% of the reference rate (Crouch et al., 1978).
Catalase (CAT) activity was measured in supernatants (1.0 mg protein/mL). Supernatant (10 µL) was mixed with reaction medium (290 µL, 20 mM H2O2, 50 mM Tris-base, 0.25 mM EDTA, pH 8.0, 25°C) in UV-star™ 96-well microplates (Greiner Bio-One) and absorbance decrease was immediately measured at 240 nm for 1 min. Molar extinction coefficient for H2O2 of 40 M− 1.cm− 1 was used to calculate catalase activity (Aebi, 1974).
Glutathione S-transferase (GST) was measured in supernatants (1.0 mg protein/mL). Supernatant (50 µL) was placed in 96-well microplate, immediately followed by reaction medium (100 µL, 1.5 mM GSH (glutathione), 2.0 mM CDNB (1-chloro-2,4-dinitrobenzene), 0.1 M potassium phosphate buffer, pH 6.5). Absorbance increase was measured at 340 nm for 2 min and the molar extinction coefficient for CDNB of 9.6 mM− 1.cm− 1 was used to calculate the activity (Keen et al., 1976).
Lipid peroxidation (LPO) was measured by Ferrous Oxidation-Xylenol (FOX) assay (Jiang et al., 1992). From the cell homogenates, 50 µL were separated for protein quantification and to the remaining volume (450 µL), an equal volume of methanol was added. After centrifugation (1,000 g for 10 min at 4°C), 30 µL of supernatant and 270 µL reaction solution (0.1 mM xylenol orange, 25 mM H2SO4, 4.0 mM BHT (butylated hydroxytoluene) and 0.25 mM FeSO4.NH4 (ammonium ferrous sulfate) in 90% methanol) were mixed in a 96-well microplate. Samples were incubated at room temperature for 30 min, centrifuged (1,000 g, room temperature for 5 min), 250 µL of supernatant were added to 96-well microplates and absorbance was measured at 570 nm. To determine the hydroperoxides concentration, the apparent molar extinction coefficient for H2O2 and cumene hydroperoxide of 4.3x104 M− 1.cm− 1 was used.
2.4) Histological analysis
The lung was chemically fixed in paraformaldehyde solution (4%, pH 7.2–7.4 for 4 h), washed in ethanol (70%), dehydrated in ethanol graded series, diaphanized in xylol and embedded in Paraplast Plus resin (Merck® - Darmstadt, Germany). Lung samples (n = 5/group) were cut in microtome (5 µm thickness) and stained with hematoxylin-eosin. The images were obtained using motorized Axio Imager Z2 epifluorescence microscope (Carl Zeiss, Jena, DE), equipped with an automated scanning Metafer 4/VSlide (Metasystems, Altlussheim, DE) and a conventional light microscope for metastasis analysis.
2.5) Integrated response of biochemical biomarkers (IBR)
The data from biomarkers corresponding to the tested doses and the control group were first transformed to logarithm to reduce the variance. These data (Yi), the mean (µ) and the standard deviation (s) were calculated from all analyzed samples. The formula Zi = (Yi - µ)/s was applied for each treatment, and the difference between the treated and control group was used to obtain the value of A (corresponding to the integrated result for each biomarker) (Beliaeff and Burgeot, 2002; modified by Sanchez et al., 2013). Then, these results were plotted on a radar-type graph, where values above and below zero (control) indicate the stimulus and inhibition of a given biomarker, respectively. Finally, to calculate the IBR index from each experimental group, the values of A were converted in absolute numbers (S) and summed.
2.6) Bioinformatic analyses - molecular docking and metabolism prediction
Dacarbazine and BDE-209 are metabolized mainly in the liver, primarily by cytochrome P450 isoform CYP1A2 (Letcher et al., 2014; Pourahmad et al., 2009). In this sense, molecular docking analysis was performed to assess the potential binding of BDE-209 and Dacarbazine at the CYP1A2 binding site as an in-silico alternative to show a possible model of how BDE-209 can interfere with dacarbazine metabolism. The 3D structure of BDE-209 was obtained from PubChem and the crystallographic structure of CYP1A2 from the Protein Data Bank (ID: 2HI4; Resolution: 1.95 Å). The files were prepared for the docking procedure in the AutoDock Tools software using a specific site docking strategy (Coordinate Grid - x: 30; y: 30; z: 30) and the LGA (Lamarckian Genetic Algorithm) algorithm to search the best ligand conformation, following the exact protocol proposed by Forli et al. (2016). Each ligand/protein pair was docked in triplicate to calculate the average affinity score (Kcal/mol) of protein-ligand complex. Finally, the protein-ligand complex was analyzed in the program Chimera 1.14 (Petersen et al., 2004) and Discovery Studio Visualizer (Biovia) in order to obtain the 3D and 2D model of the protein-ligand interaction, respectively. In addition, the prediction of pharmacokinetic metabolism of dacarbazine and BDE-209 were evaluated by the pkCSM server, which uses graph-based signatures to develop predictive models of central ADMET properties based on SMLIES chemical sequence (http://structure.bioc.cam.ac.uk/pkcsm) (Pires et al., 2015). Metabolism parameters were evaluated for inhibition of the P450 complex (CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4) and P-glycoprotein inhibitor/substrate.
2.7) Statistical procedures
The control and vehicle groups were compared using the t-test revealing no statistical differences, and so only the vehicle group is shown in the results. For all comparisons, two-way ANOVA was performed, considering sex and treatment, followed by Tukey’s post-tests. Between sexes, to compare all treatments related to the control/vehicle group, one-way ANOVA was performed with Tukey’s as posteriori test. To answer the effects of dacarbazine the Dunns’s posterior test was performed comparing the groups dacarbazine, BDE-209 and BDE-209 + dacarbazine.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}