Overexpression of FOXC1 downregulates CTH expression and increases ROS levels.
To explore whether FOXC1 participates in the regulation of amino acid metabolism in HCC cells, we performed an amino acid metabolism RT2 Profiler PCR array to examine transcriptome changes mediated by FOXC1 overexpression in Huh7 cells (Supplementary TableS1) and FOXC1 down-expression in MHCC97H cells (Supplementary TableS2). Using twofold as a cut-off to designate differentially expressed genes, 21 out of the 158 amino acid metabolism genes were down-regulated in Huh7 cells which overexpressed FOXC1, while 30 genes were up-regulated in MHCC97H cells which down-expressed FOXC1. Among the overlap of down-regulated genes in Huh7-FOXC1 and up-regulated genes in MHCC97H-shFOXC1 cells (Fig. 1A), CTH attracted our attention, which is strongly inhibited by FOXC1 overexpression. CTH is the key enzyme for cysteine synthesis (Fig. 1B) and cysteine is the precursor of ROS scavenger(23). We found that the levels of ROS increased in HCC cells with FOXC1 overexpression and decreased when FOXC1 is down-regulated (Fig. 1C). Moreover, when FOXC1 overexpressed, the ratio of glutathione (GSH)/ Glutathione (Oxidized) (GSSG) and the level of cysteine and GSH decreased. Correspondingly, the ratio of GSH/GSSG and the level of cysteine and GSH increased when FOXC1 is down-regulated (Fig. 1D).
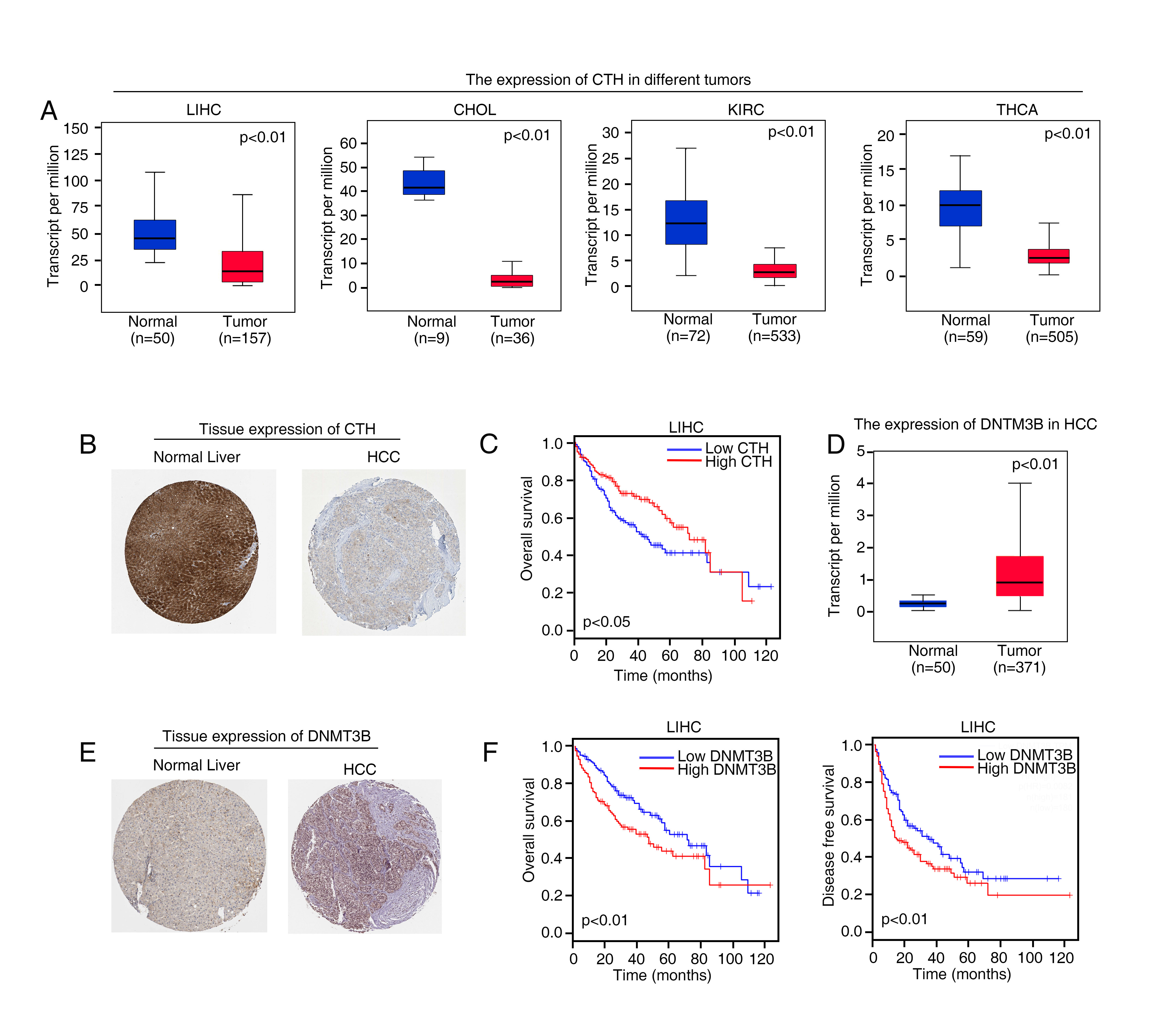
According to the data from The Cancer Genome Atlas dataset (TCGA), we found that compared with mRNA level of the CTH in normal liver tissues, the mRNA levels of CTH was markedly lessened in HCC specimens. Furthermore, based on TCGA data, the mRNA expression of CTH decreased in cholangiocarcinoma (CHOL), kidney renal clear cell carcinoma (KIRC) and thyroid carcinoma (THCA) tissues (Supplementary Figure S1A). Human Protein Atlas program database showed high or medium CTH staining intensity in normal liver samples, whereas liver cancer samples showed low staining of CTH by immunohistochemistry (IHC) tissue microarray data (Supplementary Figure S1B). Kaplan-Meier analysis based on TCGA data showed that HCC patients who had low CTH mRNA level, their overall survival time of were significantly shorter than that with high CTH mRNA level (Supplementary Figure S1C). These studies suggested that low levels of CTH indicated poor prognosis, and CTH may act as an important tumor suppressor gene (TSG) in human HCC.
FOXC1 facilitates HCC proliferation and metastasis by inhibiting CTH expression.
To explore whether FOXC1 regulates CTH expression, we first detected CTH level in four stable cell lines, Huh7-FOXC1 and MHCC97H-shFOXC1 and their control groups. Up-regulation of FOXC1 expression meaningfully inhibited the expression of CTH, whereas down-regulation of FOXC1 increased the CTH levels (Fig. 1D). We upregulated the expression of CTH in Huh7-FOXC1 cells and knocked down CTH in MHCC97H-shFOXC1 cells with lentivirus transfection (Fig. 1E). Up-regulation of CTH abolished FOXC1-facilitated cell proliferation, migration and invasion, and konckdown of CTH had the opposite results (Fig. 1F-J).
Up-regulation of CTH significantly inhibited tumor growth induced by Huh7-FOXC1 cells, whereas down-regulated CTH rescued the decreased tumor growth mediated by FOXC1 knockdown by In vivo tumorigenicity assays (Fig. 1K-L). Consistently, in vivo metastasis assay indicated that overexpression of CTH reduced the HCC metastasis with Huh7-FOXC1 cells, which prolonged overall survival time. In contrast, the down-regulation of CTH recused the inhibition of HCC proliferation and metastasis in the MHCC97H-shFOXC1 group (Fig. 1M-Q). These studies showed that FOXC1 facilitated HCC proliferation and metastasis via inhibiting CTH expression.
FOXC1 upregulates DNMT3B expression, which results in the DNA hypermethylation of CTH promoter and CTH gene silencing.
Epigenetic modifications such as DNA methylation of TSG promoters contribute to the progression and metastasis of HCC(24). Recent studies indicated that the inhibition of CTH gene transcription resulted from DNA hypermethylation of CpG rich region in the CTH promoter(25–27). To determine whether promoter DNA hypermethylation results in CTH gene silencing in FOXC-overexpressing HCC cells, the methylation status of CpG island in the CTH promoter was analyzed by bisulfite genome sequencing (BGS) analysis, which covered 13 CpG sites from − 206 to -20 of the CTH promoter (Fig. 2A). Hypermethylation at CpG sites in the CTH promoter region was detected in Huh7-FOXC1 cells, whereas less methylation was detected in MHCC97H-shFOXC1 (Fig. 2A and 2B). Compared with adjacent nontumor tissues, HCC tissues showed much higher methylation levels at CpG sites in the CTH promoter region (Fig. 2C). In addition, treatment with a demethylation agent, the DNA methyltransferase inhibitor 5-aza-2’-deoxycytidine (5-Aza), restored CTH expression in Huh7-FOXC1 cells, indicating that promoter DNA hypermethylation contributes to the transcriptional silencing of CTH gene (Fig. 2D).
As the important DNA methyltransferases, DNMT3A and DNMT3B are involved in de novo methylation patterns, which are maintained by DNMT1(28). Therefore, the DNA hypermethylation of CTH promoter may be induced by DNMTs. We found that the expression of DNMT3B is increased in Huh7-FOXC1 cells, whereas DNMT3B expression is downregulated in MHCC97H-shFOXC1 cells. However, the expression of DNMT1 and DNMT3A had no significant change in Huh7-FOXC1 and MHCC97H-shFOXC1 cells compared to control cells (Fig. 2E). Moreover, ectopically overexpression of DNMT3B decreased the expression of CTH in MHCC97H-shFOXC1 cells, whereas knockdown of DNMT3B increased CTH expression in Huh7-FOXC1 cells (Fig. 2F). These studies suggested that FOXC1 upregulated DNMT3B expression, which resulted in CTH gene silencing in HCC cells.
DNMT3B is a direct transcriptional target of FOXC1.
The mechanism by which FOXC1 upregulated DNMT3B expression is still unclear. We hypothesized that FOXC1 may directly transactivate DNMT3B. A dual-luciferase reporter assay showed that overexpression of FOXC1 enhanced the transcription of the luciferase reporter in the DNMT3B plasmid constructs compared to that of the controls (Fig. 2G). In order to clarify the regulation mechanism of DNMT3B, the promoter sequence of DNMT3B was analyzed and we found 3 putative FOXC1 binding motifs. To further confirm whether FOXC1 targets these potential binding sites to regulate DNMT3B, we designed a series of reporter plasmids containing truncated or mutated DNMT3B promoter sequences and used them in luciferase reporter assay. Huh7 cells were transfected with these plasmids to assess their reaction to FOXC1 overexpression. The results suggested that FOXC1-induced luciferase reporter expression was significantly eliminated by deletion in the − 195 ~ + 109 bp region compared to that of the controls. Consistently, compared with the control group, mutations at the putative binding sites 2&1 of the DNMT3B promoter significantly reduced the activity of the FOXC1 overexpressed luciferase reporter gene, suggesting that these binding sites were essential for FOXC1 transactivation (Fig. 2H). A chromatin immunoprecipitation (ChIP) assay demonstrated the direct binding of FOXC1 to the putative site in the DNMT3B promoter in Huh7-FOXC1 cells (Fig. 2I upper). Moreover, we investigated whether FOXC1 binds to the same regions in patient samples, and the results revealed that compared to healthy controls, FOXC1 binding sites in the HCC samples were indeed enriched in these regions. (Fig. 2I, lower).
Then we examined the protein levels of CTH and promoter methylation levels of CTH in two independent cohorts of human HCC tissues. Microvascular invasion, poor tumor differentiation, and higher TNM stage were positively associated with the depletion of CTH (Table 1). Loss of CTH expression was an independent and meaningful risk factor for recurrence and reduced survival according to the multivariate analysis (Table 2). The expression of CTH was inversely correlated with promoter methylation levels of CTH in both cohorts (Fig. 2J). In addition, promoter methylation of CTH gene was positively correlated with aggressive tumor behaviors (Supplementary Table S3). HCC patients with promoter methylation of CTH had higher recurrence rates and shorter overall survival time than HCC patients without promoter methylation of CTH (Fig. 2K).
Table 1
Correlation between CTH expression and clinicopathological characteristics of HCCs in two independent cohorts of human HCC tissues
| | | Cohort I | | | Cohort II | |
| Clinicopathological variables | Tumor CTH expression | P Value | | Tumor CTH expression | P Value |
| Negative (n = 152) | Positive (n = 128) | | Negative (n = 109) | Positive (n = 101) |
| Age | 51.63(9.148) | 52.85(10.966) | 0.283 | | 50.95(10.857) | 54.59(9.943) | 0.546 |
| Sex | female | 25 | 20 | 0.872 | | 20 | 19 | 1.000 |
| | male | 127 | 108 | | | 89 | 82 | |
| Serum AFP | ≤ 20 ng/ml | 24 | 25 | 0.433 | | 19 | 31 | 0.034 |
| | > 20 ng/ml | 128 | 103 | | | 90 | 70 | |
| Virus infection | HBV | 108 | 85 | 0807 | | 76 | 82 | 0.129 |
| | HCV | 22 | 22 | | | 11 | 8 | |
| | HBV + HCV | 9 | 7 | | | 5 | 5 | |
| | none | 13 | 14 | | | 17 | 6 | |
| Cirrrhosis | absent | 38 | 41 | 0.230 | | 28 | 29 | 0.644 |
| | present | 114 | 87 | | | 81 | 72 | |
| Child-pugh score | Class A | 122 | 114 | 0.049 | | 81 | 77 | 0.752 |
| | Class B | 30 | 14 | | | 28 | 24 | |
| Tumor number | single | 91 | 100 | 0.001 | | 56 | 68 | 0.024 |
| | multiple | 61 | 28 | | | 53 | 33 | |
| Maximal tumor size | ≤ 5 cm | 78 | 84 | 0.021 | | 45 | 54 | 0.097 |
| | > 5 cm | 74 | 44 | | | 64 | 47 | |
| Tumor encapsulation | absent | 54 | 21 | < 0.001 | | 62 | 24 | < 0.001 |
| | present | 98 | 107 | | | 47 | 77 | |
| Microvascular invasion | absent | 81 | 91 | 0.003 | | 41 | 75 | < 0.001 |
| | present | 71 | 37 | | | 68 | 26 | |
| Tumor differentiation | I-II | 100 | 107 | 0.001 | | 76 | 91 | <0.001 |
| | III-Ⅳ | 52 | 21 | | | 33 | 10 | |
| TNM stage | I-II | 101 | 122 | < 0.001 | | 73 | 97 | < 0.001 |
| | III | 51 | 6 | | | 36 | 4 | |
Table 2
Univariate and Multivariate Analysis of Factors Associated with Time To Recurrence and Overall Survival in Cohort I HCC Patients (n = 280) and Cohort II HCC Patients (n = 210)
| Cohort I (n = 280) | Time To Recurrence | | Overall Survival |
| Clinical Variables | HR(95%CI) | P value | | HR(95%CI) | P value |
| Univariate Analysis | | | | | |
| Age | 0.994(0.979–1.009) | 0.427 | | 0.989(0.973–1.004) | 0.152 |
| Sex (female versus male) | 0.861(0.574–1.293) | 0.861 | | 0.902(0.592–1.373) | 0.630 |
| Serum AFP (≤ 20 versus > 20 ng/ml) | 1.418(0.927–2.170) | 0.108 | | 1.301(0.849–1.994) | 0.227 |
| Virus infection (no versus yes) | 0.986 (0.796–1.222) | 0.896 | | 1.002(0.808–1.243) | 0.986 |
| Cirrhosis ( absent versus present) | 1.039(0.741–1.456) | 0.825 | | 1.132(0.797–1.606) | 0.489 |
| Child-pugh score (A versus B) | 1.254(0.835–1.884) | 0.274 | | 1.247(0.824–1.887) | 0.296 |
| Tumor number (single versus multiple) | 2.596(1.903–3.540) | < 0.001 | | 0.343(0.250–0.470) | < 0.001 |
| Maximal tumor size (≤ 5 versus > 5 cm) | 0.583(0.482–0.706) | 0.013 | | 0.696(0.509–0.951) | 0.023 |
| Tumor encapsulation (absent versus present) | 0.341(0.248–0.469) | < 0.001 | | 3.065(2.221–4.230) | < 0.001 |
| Microvascular invasion (absent versus present) | 2.338(1.720–3.179) | < 0.001 | | 0.405(0.296–0.554) | < 0.001 |
| Tumor differentiation (I-II versus III-Ⅳ) | 0.295(0.241–0.361) | < 0.001 | | 0.317(0.229–0.439) | < 0.001 |
| TNM stage (I-II versus III) | 3.032(2.193–4.191) | < 0.001 | | 0.145(0.102–0.205) | < 0.001 |
| CTH (negative versus positive) | 6.289(4.444–8.901) | < 0.001 | | 3.733(2.629–5.299) | < 0.001 |
| Multivariate analysis | | | | | |
| Tumor number (single versus multiple) | 0.707(0.473–1.057) | 0.091 | | 0.616(0.413–0.920) | 0.018 |
| Maximal tumor size (≤ 5 versus > 5 cm) | 1.050(0.749–1.471) | 0.778 | | 1.085(0.766–1.537) | 0.645 |
| Tumor encapsulation (absent versus present) | 1.448(0.937–2.238) | 0.096 | | 1.392(0.889–2.178) | 0.148 |
| Microvascular invasion (absent versus present) | 0.674(0.469–0.968) | 0.033 | | 0.631(0.436–0.912) | 0.014 |
| Tumor differentiation (I-II versus III-Ⅳ) | 0.858(0.514–1.435) | 0.560 | | 0.933(0.548–1.587) | 0.798 |
| TNM stage (I-II versus III) | 0.357(0.197–0.645) | 0.001 | | 0.335(0.184–0.611) | < 0.001 |
| CTH (negative versus positive) | 2.759(1.941–3.922) | < 0.001 | | 2.969(2.054–4.293) | < 0.001 |
| Cohort II (n = 210) | Time To Recurrence | | Overall Survival |
| Clinical Variables | HR(95%CI) | P value | | HR(95%CI) | P value |
| Univariate Analysis | | | | | |
| Age | 0.987(0.970–1.004) | 0.122 | | 0.985(0.968–1.002) | 0.076 |
| Sex (female versus male) | 0.769(0.499–1.184) | 0.232 | | 0.722(0.468–1.115) | 0.722 |
| Serum AFP (≤ 20 versus > 20 ng/ml) | 1.199(0.788–1.825) | 0.397 | | 1.248(0.809–1.927) | 0.316 |
| Virus infection (no versus yes) | 0.959 (0.700-1.313) | 0.792 | | 0.923(0.669–1.275) | 0.628 |
| Cirrhosis ( absent versus present) | 0.873(0.595–1.282) | 0.489 | | 0.860(0.583–1.271) | 0.450 |
| Child-pugh score (A versus B) | 0.973(0.649–1.460) | 0.896 | | 0.981(0.649–1.482) | 0.927 |
| Tumor number (single versus multiple) | 1.984(1.398–2.817) | < 0.001 | | 0.484(0.338–0.691) | < 0.001 |
| Maximal tumor size (≤ 5 versus > 5 cm) | 1.328(0.934–1.888) | 0.114 | | 0.705(0.491–1.012) | 0.058 |
| Tumor encapsulation (absent versus present) | 0.354(0.249–0.505) | < 0.001 | | 3.085(2.148–4.431) | < 0.001 |
| Microvascular invasion (absent versus present) | 2.319(1.629–3.301) | < 0.001 | | 0.389(0.271–0.559) | < 0.001 |
| Tumor differentiation (I-II versus III-Ⅳ) | 2.128(1.422–3.184) | < 0.001 | | 0.431(0.287–0.647) | < 0.001 |
| TNM stage (I-II versus III) | 7.507(4.967–11.345) | < 0.001 | | 0.124(0.082–0.188) | < 0.001 |
| CTH (negative versus positive) | 0.261(0.178–0.382) | < 0.001 | | 4.409(2.950–6.588) | < 0.001 |
| Multivariate analysis | | | | | |
| Tumor number (single versus multiple) | 1.011(0.667–1.534) | 0.958 | | 0.986(0.638–1.523) | 0.949 |
| Maximal tumor size (≤ 5 versus > 5 cm) | 0.856(0.510–1.436) | 0.556 | | 0.787(0.462–1.340) | 0.377 |
| Tumor encapsulation (absent versus present) | 1.679(1.012–2.786) | 0.045 | | 1.636(0.976–2.742) | 0.062 |
| Microvascular invasion (absent versus present) | 1.111(0.660–1.871) | 0.693 | | 1.000(0.591–1.694) | 1.000 |
| Tumor differentiation (I-II versus III-Ⅳ) | 0.851(0.547–1.324) | 0.475 | | 0.778(0.499–1.212) | 0.267 |
| TNM stage (I-II versus III) | 0.173(0.094–0.321) | 0.001 | | 0.172(0.091–0.323) | < 0.001 |
| CTH (negative versus positive) | 3.403(2.243–5.164) | < 0.001 | | 3.816(2.467–5.903) | < 0.001 |
DNMT3B is critical for FOXC1-induced HCC proliferation and metastasis.
To explore whether DNMT3B was involved in FOXC1-mediated HCC proliferation and metastasis, we knocked down DNMT3B in Huh7-FOXC1 cells and ectopically upregulated DNMT3B expression in MHCC97H-shFOXC1 cells with lentivirus transfection (Fig. 2F). Down-regulation of DNMT3B inhibited FOXC1-facilitated HCC proliferation, migration, and invasion abilities, while up-regulation of DNMT3B had the opposite result (Fig. 3A-E). In vivo tumorigenicity assays suggested that down-regulated DNMT3B significantly inhibited tumor growth induced by Huh7-FOXC1 cells (Fig. 3F, left), whereas upregulation of DNMT3B salvaged the suppression tumor growth induced by FOXC1 knockdown (Fig. 3F, right). Representative Ki67-stained images are shown (Fig. 3G). In vivo metastatic assay suggested that sown-regulated DNMT3B eliminated HCC metastasis with Huh7-FOXC1 cells, which prolonged overall survival time. In contrast, overexpression of DNMT3B rescued the inhibition of HCC metastasis in the MHCC97H-shFOXC1 groups (Fig. 3H-K). Representative H&E-stained images are shown (Fig. 3L). These results identified that FOXC1 facilitated HCC proliferation and metastasis through upregulating DNMT3B expression.
FOXC1 expression is positively correlated with DNMT3B expression and negatively correlated with CTH expression in human HCC tissues.
Immunohistochemical (IHC) analysis was used to verify the clinical relevance of FOXC1 and DNMT3B or CTH in human HCC specimens from two independent cohorts. The representative images of FOXC1, DNMT3B, and CTH expression were showed by IHC staining (Fig. 4A). Compared to adjacent nontumor tissues, DNMT3B expression was observably increased in HCC tissues, whereas CTH expression was observably decreased in HCC tissues. FOXC1 expression was positively associated with DNMT3B expression but negatively associated with CTH expression in both cohorts (Fig. 4B). Up-expression of DNMT3B was positively correlation with poor prognosis (Fig. 4C and 4E, upper panel) and aggressive tumor behavior (Supplementary TableS4). Both TCGA database and Human Protein Atlas program database showed that compared to the normal liver tissues, the mRNA and protein levels of DNMT3B in liver cancer tissues were much higher (Supplementary Figure S1D-E). Kaplan–Meier analysis based on TCGA data displayed that compared to HCC patients with low DNMT3B mRNA levels, patients who have high DNMT3B mRNA levels had shorter overall survival time and disease-free survival time (Supplementary Figure S1F). In addition, the reduced expression of CTH indicated poor prognosis (Fig. 4D and 4F, upper panel). Patients who had both of high expression of FOXC1 and DNMT3B endured the highest recurrence rates and lowest overall survival time (Fig. 4C and 4E, lower panel). Consistently, patients with the FOXC1(+)/CTH (-) expression pattern had the highest recurrence rates and lowest overall survival time (Fig. 4D and 4F, lower panel).
High levels of ROS are critical for FOXC1-mediated HCC cell proliferation, migration and invasion.
To explore whether ROS regulate FOXC1-mediated HCC proliferation, migration and invasion, N-acetylcysteine (NAC) which is an antioxidant(29) and L-Buthionine-sulfoximine (BSO) which decreases GSH levels(30), were used to treat Huh7 -FOXC1 cells and MHCC97H-shFOXC1 cells, respectively. NAC (0.2 mM) treatment abolished high levels of ROS induced by FOXC1 overexpression, whereas BSO (30 µM) treatment rescued the decreased ROS levels induced by FOXC1 knockdown (Fig. 5A). Meanwhile, low levels of ROS induced by NAC inhibited the cell proliferation, migration, and invasion abilities mediated by FOXC1 overexpression and high levels of ROS induced by BSO rescued the suppression results mediated by FOXC1 knockdown (Fig. 5B-E).
High Levels of ROS upregulates FOXC1 expression through ERK1/2-pELK1 pathway.
We have elucidated that overexpression of FOXC1 increased ROS levels by inhibiting cysteine metabolism and ROS promotes HCC progression and metastasis. Meanwhile, we previously reported that FOXC1 is over-expressed in human HCC tissues and promoted HCC progression and metastasis(21, 22). Therefore, we determined whether high levels of ROS regulate FOXC1 expression in HCC cells. Treatment with BSO and NAC could increase and decrease ROS levels respectively (Fig. 5F). BSO treatment significantly increased FOXC1 expression and NAC treatment decreased FOXC1 expression at protein level in HCC cells (Fig. 5G). Notably, with the stimulation of BSO, FOXC1 promoter activity was significantly increased, indicating that high levels ROS transactivated FOXC1 promoter to elevated the expression of FOXC1 (Fig. 5H).
To verify the accurate location of cis-regulatory elements in the FOXC1 promoter sequence that reacted to ROS, we generated a series of truncated mutants of the FOXC1 promoter (Fig. 5I). Dramatic suppression in FOXC1 promoter activity were found in mutants with two deletions from nt-1058 to nt-422 and nt-422 to nt-66, suggesting that these sequences are important in allowing ROS-enhanced FOXC1 activation. The prior region contains one ELK1 binding site, one specificity protein 1 (SP1) binding site and one ATF2 binding site. And the later region contains one SP1 binding site and ELK1 binding site. Notably, site-directed mutagenesis at the ELK1 binding sites inhibited the ROS-promoted FOXC1 activity, while no effect was found for mutations at the SP1 binding site and ATF2 binding site (Fig. 5I). Meanwhile, knockdown of ELK1 abolished FOXC1 promoter activity which is increased by BSO (Fig. 5J). We found that BSO didn’t increase the expression of ELK1 but activate phosphorylation of ELK1 when activated FOXC1 expression (Fig. 5K).
ROS activates Nuclear factor kB (NF-kB), c-Jun N-terminal kinase (JNK), ERK1/2, p38 kinases and Phosphoinositide 3-kinase (PI3K)/ protein kinase B (Akt) pathways to promote cancer progression(31). To verify that ROS regulate FOXC1 expression via which pathway, we treated cells with PI3K, p38 kinases, JNK, NF-κB and ERK1/2 inhibitors. Preconditioning cells with ERK1/2 inhibitor decreased ROS-induced FOXC1 expression (Fig. 5L). Nevertheless, there was on effect on ROS regulating FOXC1 expression when cells were pretreated with other inhibitors. Further, ChIP assays showed that the ERK1/2 inhibitor dramatically weakened ELK1 binding to the FOXC1 promoter, while there were on significant changes on the binding of ELK1 to the FOXC1 promoter by other inhibitors (Fig. 5M). These results demonstrated that ROS induced FOXC1 over-expression via the ERK1/2-p-ELK1 signaling pathway.
8-OHdG is the oxidative damage marker(32). IHC was used to detect 8-OHdG, phospho-ELK1 (activated ELK1) expression in two independent cohorts of human HCC tissue arrays. Compared to adjacent nontumor tissues, both 8-OHdG and p-ELK1 expression was markedly elevated in HCC tissues. 8-OHdG and p-ELK1 expression were localized in the nucleus (Fig. 6A). FOXC1 expression was positively associated with both 8-OHdG and p-ELK1 expression in two cohorts (Fig. 6B). patients with high expression of 8-OHdG (Fig. 6C and 6E, upper panel) and p-ELK1(Fig. 6D and 6F, upper panel), compared to patients with low expression of 8-OHdG and p-ELK1, had shorter overall survival and higher recurrence rates. Elevated expression of both 8-OHdG and p-ELK1 were positively correlated with loss of tumor encapsulation, microvascular invasion, poor tumor differentiation, and a higher TNM stage (Supplementary TableS5-S6). Furthermore, patients with positive co-expression of 8-OHdG (Fig. 6C and 6E, lower panel) and FOXC1 or co-expression of p-ELK1 ((Fig. 6D and 6F, upper panel)) and FOXC1 had the highest recurrence rates and lowest overall survival times.
{kind=link}