The chemicals and reagents were acquired from Sigma Aldrich, Himedia, and SD Fine Chemicals, Mumbai, India. The homogeneity of the compounds was carried out with TLC pre-coated Silica GF254 plates employing ethyl acetate and n-hexane (4:6) as a solvent system and visualization of TLC spots was carried out in the UV chamber. Melting points were carried out in Equiptronics digital melting point apparatus and are uncorrected. FT-IR spectrum (cm-1) was recorded on an Alpha Bruker IR spectrometer. Proton spectra (1H NMR) and C13NMR were recorded on the Agilent 400MR DD2 FT-NMR spectrophotometer operating at 400MHz. TMS is used as an internal standard (δ ppm). Waters HR-MS mass spectrometer operating at 70ev employing ESI and TOF detector, mass spectra were recorded.
Synthesis of Dithiocarbamate
Step-(1): An equimolar solution of sodium hydroxide (0.05 mol) and 2- Aninobenzothiazole (0.05 mol) was stirred together. CS2 (8ml) was gradually introduced to the above solution, accompanied by stirring for 15-20 min, followed by additional stirring at 0-5°C for 3hr to yield the corresponding dithiocarbamate. The mixture was then stirred at room temperature, and the precipitate was filtered, washed, and dried. Recrystallization was done using ethanol.
Preparation of sodium salt of chloroacetic acid solution
Equimolar solution of Sodium hydroxide (0.05 mol) and Monochloroacetic acid (0.05 mol) was taken in a beaker, mixed, and stirred for 30 min at 0-5°C. The solution is converted basic to litmus paper by the addition of sodium carbonate.
Procedure for the synthesis of Benzothiazole incorporated N-substituted rhodanines
Step-(2): A solution of freshly prepared sodium chloroacetate was introduced to the solution containing dithiocarbamate (1) (0.01 mol) with stirring at 0-5°C. The stirring was further continued for 3 hr, followed by cyclization with concentrated HCl (65-70°C, 30 min). The formation of the product was monitored with the help of thin layer chromatography (TLC). The reaction mixture is allowed to cool at RT, followed by filtration and recrystallization using ethanol.
General procedure for the synthesis of benzothiazole substituted rhodanines by Knoevenagel condensation (A1-10)
N-substituted rhodanines (2) (0.01 mol) and aromatic aldehydes (0.01 mol) were dissolved in 15-20 ml of toluene in a 500ml round bottom flask. Piperidine and glacial acetic acid were added in catalytic amounts and the contents were refluxed for 8-10 hrs. (100-110ºC). The formation of the product was monitored with the help of TLC and followed by cooling the reaction mixture. The precipitated compound is filtered, washed with water, and recrystallization was done using ethanol [15]. The physical data of compounds (A1-10) is given in Table-1.
(Z)-3-(benzo[d]thiazol-2-yl)-5-(4-nitrobenzylidene)-2 thioxothiazolidin-4-ones (A1): Yellow solid. Yield: 67%, melting point (MP). 233-35°C, FT-IR (cm-1): 3065 (Ar,C-H), 1700 (C=O), 1611 (C=N), 1531(C=C). 1H-NMR (400 MHz, DMSO-d6, δ, ppm): 7.228-7.963 (m, Ar, 8H), 8.03(s, =CH, 1H). 13CNMR (400MHz, DMSOd6, δ, ppm): 113.42, 115.97, 118.13, 118.23, 122.05, 125.23, 126.23, 127.34, 140.01, 143.82, 145.78, 154.01, 155.89, 157.78, 162.71, 176.10 and 202.24. HR-MS (m/z): calculated for C17H9N3O3S3 is 398.98; found: 398.9896 (M+).
(Z)-3-(benzo[d]thiazol-2-yl)-5-(4-bromobenzylidene)-2-thioxothiazolidin-4-one (A2): Yellow solid. Yield 72%, MP. 245-47°C, FT-IR (cm-1): 3048 (Ar, C-H), 1725 (C=O), 1655 (C=N), 1593 (C=C). 1H-NMR (400 MHz, DMSO-d6, δ, ppm): 6.98-7.51 (m, Ar, 8H), 7.61 (s, =CH, 1H). 13C-NMR (400MHz, DMSO-d6, δ, ppm): 111.16, 115.84, 116.53, 120.50, 121.56, 121.85, 123.00, 125.39, 126.47, 129.10, 131.24, 131.60, 131.79, 148.47, 154.10, 164.00 and 192.02. HR-MS (m/z): calculated for C17H9BrN2OS3 is 433.91; found: 433.9170(M+).
(Z)-3-(benzo[d]thiazol-2-yl)-5-(4-chlorobenzylidene)-2-thioxothiazolidin-4-one (A3): Yellow solid. Yield 69%, MP. 273-75°C, FT-IR (cm-1): 3048 (Ar, C-H), 1655 (C=O), 1593 (C=N), 1516 (C=C). 1H-NMR (400 MHz, DMSO-d6, δ, ppm): 6.97 - 7.62 (m, Ar, 8H), 7.6 (s, =CH, 1H). 13C-NMR (400MHz, DMSO-d6, δ, ppm): 115.11, 117.68, 118.11, 119.91, 121.29, 122.38, 122.71, 123.42, 125.87, 126.05, 132.32, 132.41, 142.13, 146.47, 153.52, 179.51 and 196.23. HR-MS (m/z): calculated for C17H9ClN2OS3 is 354.47; found: 354.4713 (M+).
Z)-3-(benzo[d]thiazol-2-yl)-5-benzylidene-2-thioxothiazolidin-4-one (A4): Yellow solid. Yield 65%, MP. 259-61°C; FT-IR (cm-1): 3010 (Ar, C-H), 1723 (C=O), 1631(C=N), 1531 (C=C). 1H-NMR (400 MHz, DMSO-d6, δ, ppm): 6.97 - 7.47 (m, Ar, 8H), 7.63 (s, =CH, 1H). 13C-NMR (400MHz, DMSO-d6, δ, ppm): 116.32, 117.68, 118.11, 119.91, 121.29, 122.38, 122.71, 123.42, 125.87, 126.05, 132.32, 132.41, 142.13, 146.47, 153.52, 169.43 and 196.23. HR-MS (m/z): calculated for C17H10N2OS3 is 354.47; found: 354.47 (M+).
(Z)-3-(benzo[d]thiazol-2-yl)-5-(4-hydroxy-3-methoxybenzylidene)-2-thioxothiazolidin-4-one (A5): Yellow solid. Yield 87%, MP. 214-16°C, FT-IR (cm-1): 3176 (OH), 3064 (Ar, C-H), 1722 (C=O), 1639 (C=N), 1530 (C=C). 1H-NMR (400 MHz, DMSO-d6, δ, ppm): 4.20 (s, OCH3, 3H), 6.99 - 7.53 (m, Ar-H, 7H), 7.63 (s, =CH, 1H), 9.98-9.82 (s, OH, 1H). 13C-NMR (400MHz, DMSO6, δ, ppm): 40.09, 114.47, 115.84, 117.96, 121.42, 121.47, 125.98, 130.95, 143.39, 143.84, 148.10, 150.10, 150.24, 150.24, 150.30, 150.79, 151.03 168.54 and 191.54. HR-MS (m/z): calculated: for C19H14N2O3S3 is 400.50; found: 400.5043 (M+).
(Z)-3-(benzo[d]thiazol-2-yl)-5-(3,4-dimethoxybenzylidene)-2-thioxothiazolidin-4-one (A6): Yellow solid. Yield 74%, MP. 227-29°C, FT-IR (cm-1): 3274 (OH), 3007 (Ar, C-H), 1726 (C=O), 1641 (C=N), 1460 (C=C). 1H-NMR (400 MHz, DMSO-d6, δ, ppm): 3.82 (s, OCH3, 3H), 4.26 (s, OCH3, 3H), 6.97-7.49 (m, Ar-H, 7H), 7.53 (s, =CH, 1H). 13C-NMR (400MHz, DMSOd6, δ, ppm): 55.96, 56.32, 109.92, 111.72, 118.10, 121.30, 121.59, 125.87, 126.54, 130.09, 131.26, 144.92, 149.62, 152.98, 154.64, 166.96, 168.54, 177.92 and 191.80.HR-MS (m/z): calculated for C19H14N2O3S3 is 414.52; found: 414.5277 (M+).
(Z)-3-(benzo[d]thiazol-2-yl)-5-(4-methoxybenzylidene)-2-thioxothiazolidin-4-one (A7):
Yellow solid. Yield 67%, MP. 266-68°C; FT-IR (cm-1): 3211 (OH), 3014 (Ar, C-H), 1725 (C=O), 1632 (C=N), 1531 (C=C). 1H-NMR (400 MHz, DMSO-d6, δ, ppm): 3.85 (s, OCH3, 3H), 6.99 - 7.21 (m, Ar-H, 8H), 7.31 (s, =CH, 1H). 13C-NMR (400MHz, DMSO-d6, δ, ppm): 56.12, 113.82, 114.94, 115.02, 118.14, 121.25, 121.28, 125.86, 130.10, 131.32, 132.24, 148.21, 153.16, 164.66, 166.89, 168.55, 179.92 and 191.71. HR-MS (m/z): calculated for C18H12N2O2S3 is 384.50; found: 384.5043 (M+).
(Z)-3-(benzo[d]thiazol-2-yl)-5-(4-(dimethylamino) benzylidene)-2-thioxothiazolidin-4-one (A8): Yellow solid. Yield 56%, MP. 235-37°C; FT-IR (cm-1): 2912 (Ar, C-H), 1725 (C=O), 1649 (C=N), 1529 (C=C). 1H-NMR (400 MHz, DMSO-d6, δ, ppm): 3.01 (s, 2 x CH3, 6H), 6.77 - 7.64 (m, Ar-H, 8H), 7.66 (s, =CH, 1H). 13C-NMR (400MHz, DMSO-d6, δ, ppm): 40.33, 40.53, 111.49, 118.11, 121.30, 124.95, 125.87, 131.26, 131.98, 132.24, 139.12, 140.21, 141.12, 152.99, 154.64, 154.86, 166.95, 168.53 and 190.30. HR-MS (m/z): calculated for C19H15N2OS3 is 397.54; found: 397.5439 (M+).
(Z)-3-(benzo[d]thiazol-2-yl)-5-(3, 4-dichlorobenzylidene)-2-thioxothiazolidin-4-one (A9): Yellow solid. Yield 79%, MP. 217-19°C, FT-IR (cm-1): 3194 (-OH), 2996 (Ar, C-H), 1725 (C=O), 1640 (C=N), 1532 (C=C). 1H-NMR (400 MHz, DMSO-d6, δ, ppm): 6.99 - 7.64 (m, Ar-H, 8H), 7.66 (s, =CH, 1H), 9.78 (s, OH, 1H). 13C-NMR (400MHz, DMSO-d6, δ, ppm): 114.52, 116.29, 117.86, 121.47, 121.53, 126.01, 130.77, 122.51, 130.77, 132.51, 150.14, 151.21, 154.38, 155.78, 156.15, 167.10, 168.52, 169.93 and 191.36. HR-MS (m/z): 369.96 (M+1 found) and calculated: 369.9819 (M+).
(Z)-3-(benzo[d]thiazol-2-yl)-5-(2-hydroxybenzylidene)-2-thioxothiazolidin-4-one (A10):
Yellow solid, Yield 52%, MP. 192-94°C; FT-IR (cm-1): 3293 (OH), 3064 (Ar, C-H), 1725 (C=O), 1639 (C=N), 1529 (C=C). 1H-NMR (400 MHz, DMSO-d6, δ, ppm): 6.94 - 7.49 (m, Ar-H, 8H), 7.62 (s, =CH, 1H), 10.26 (s, OH, 1H). 13C-NMR (400MHz, DMSO-d6, δ, ppm): 117.88, 118.11, 119.91, 121.29, 122.30, 122.71, 123.42, 125.87, 126.79, 129.70, 131.27, 136.82, 153.02, 161.17, 166.93, 168.53 and 192.31. HR-MS (m/z): 369.96 (M+1 found) and calculated: 369.9178 (M+).
In- silico studies:
In-silico studies were performed by determining ADMET properties, Lipinski’s rule of five, docking studies, free energy calculation, and toxicity profile, which is mainly focused to determine drug-likeness and preferred orientation of the molecule.
Drug likeness evaluation of benzothiazole‒rhodanine derivatives
Determination of Lipinski’s rule of five, physicochemical parameters ADMET properties and evaluation of toxicity of the Benzothiaozole‒Rhodanine derivatives
Lipinski’s rule of five is mainly used to verify molecular properties [19] which are essential and related to PK of the drug molecule, which is carried out with Schrodinger 2018-3 suite device Maestro 11.7.012[20]. Qikprop(https://www.schrodinger.com) is a rapid, precise, and accessible to predict ADME properties [21]. In stages of drug development, toxicity must be done and in many cases, toxic effects are identified in the later stage of the development. So in drug development, prior precise toxicity assessments are very important.
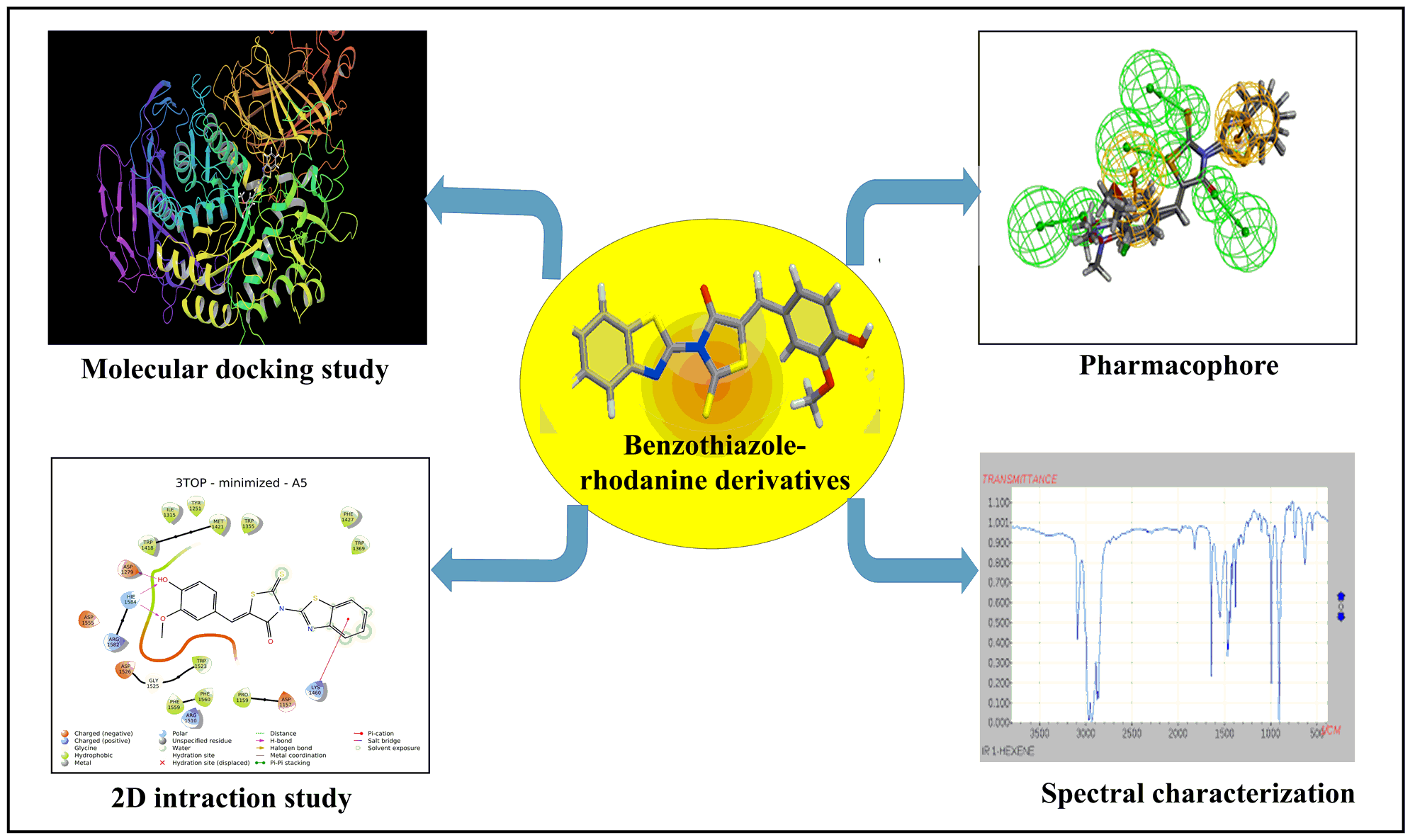
Molecular docking studies
To rationalize the results from the In-Vitro antidiabetic studies, we have further carried out flexible ligand docking using the Glide module. Molecular docking was performed by utilizing the Glide module of Schrodinger 2018-3 suite device Maestro 11.7.012 [22] (https://www.schrodinger.com) Crystal structures with good resolution of all the target proteins were taken from RCSB PDB (https://www.rcsb.org)[23]
Binding free energy (MM-GBSA) calculation
Binding free energy (MM-GBSA) calculations were performed by prime-MM-GBSA Schrodinger 2018-3 suite devices Maestro 11.7.012. Scores were taken from XP docking for free energy calculation [24,25].
Molecular dynamics (MD) simulation study
On GROMACS 5.1.5, performed with CHARMM27 all-atom force field, MD simulation analyses were carried out on native protein structures (PDB ID: 3TOP) in innate as well as docked complex form with ligand structure. Externally, all atomic force fields were generated using the SwissParam server (https://www.swissparam.ch/) using CHARMM to create the topology files for the ligand. For calculating the Van der Waal interactions, a distance cut-off of 1.0 nm was maintained. Partial Mesh Ewald (PME) summation for long-range electrostatics was applied to calculate columbic interactions (1nm cut-off).To maintain the electro-neutrality of the system, counter ions were included. Using the TIP3P water model, the system was solvated and simulations were carried out in a triclinic box wherein the protein atoms were removed from the box wall dimensions at 1.0 to 1.5 nm, keeping intact the conditions of the periodic boundary. Structure minimization was done foremost using the steepest descent algorithm and a1000 KJ/mol/nm tolerance. Position restraints were applied on the complex to equilibrate the system. Simulations were also performed by employing canonical NVT and NPT ensembles. At a temperature of 300 K and a pressure of 1 bar, both equilibria were carried out at 200 pseach. Initial velocities were yielded following Maxwell distribution and coupling of temperature was implemented using velocity rescaling (coupling constant of 0.1ps). Extended ensemble Parrinello–Rahman algorithm helped in temperature pressure coupling (coupling constant of 2 ps). The equilibrated system was further exposed to a production run of 10 ns and 2fs time step integration. XMGRACE-5.1.22 and GROMACS analysis tools program were employed to analyze the saved trajectories. (https://plasma-gate.weizmann.ac.il/Grace/).
Pharmacophore Mapping
The pharmacophore mapping identifies the molecular properties in 2D and 3D terms. The present title compounds were initially aligned to a common scaffold and prepared a dataset. The pharmacophore could be generated from several algorithms of which common feature pharmacophore application is one such. The application identifies molecular features out of the several structural properties and the individual features were kept a maximum of 2.5 Å distance with a maximum of six features as a single pharmacophore set. The best pharmacophore thus generated was selected best on fitness.
In-vitro Antidiabetic activity
α-amylase inhibitory assay
20µL of α -amylase solution (0.5 mg/ml) was added to phosphate buffer (pH 6.9, 0.02 M, 200 µL). 250 μL of test samples (10-50 µg/mL) was added, to all the above solutions and kept for incubation for 10 min.1% starch solution (200µL) was further added and again kept for incubation for 10 min at a temperature of 25°C. Termination of the reaction was affected by the addition of 400 μL of 3, 5-dinitrosalicylic acid (DNS) reagent. Lastly, it was incubated in water (at 70°C, 5 min). The absorbance was recorded using an ELISA microplate reader at 540 nm. Acarbose was served as the standard. All the reactions were carried out in triplicates. The reaction mixture, devoid of the test sample, served as control. The following formula was employed to calculate percentage inhibition. The IC50 values are tabulated in table-7[26].
% Inhibition = [(Ac – As) /Ac] × 100
Ac-Controlabsorbance, As-Standard absorbance
α-glucosidase inhibitory assay
10 µl of the α-glucosidase enzyme, 20 µl of test samples (10-50 µg/ml) and 50 µl, of 0.1M phosphate buffer (pH=6.8), were incubated at 37°C for 15 min in a 96-well plate. 20 µl of p-nitrophenyl—D-glucopyranoside solution was further added as a substrate and again incubated for 20 min at 37oC. Sodium carbonate (50 µl of 0.1M) was added to terminate the reaction. P-nitrophenol was liberated, during the reaction which was measured at 405nm using an ELISA microplate reader. Acarbose served as the standard. A control was prepared under similar conditions by omitting test samples. All the experiments were carried out in triplicates. The percentage inhibition was calculated by the formula. The IC50 values are tabulated in table-7.[26](26)
% Inhibition = [(Ac – As) /Ac] × 100
Ac-absorbance for control, As-absorbance for standard
{kind=link}