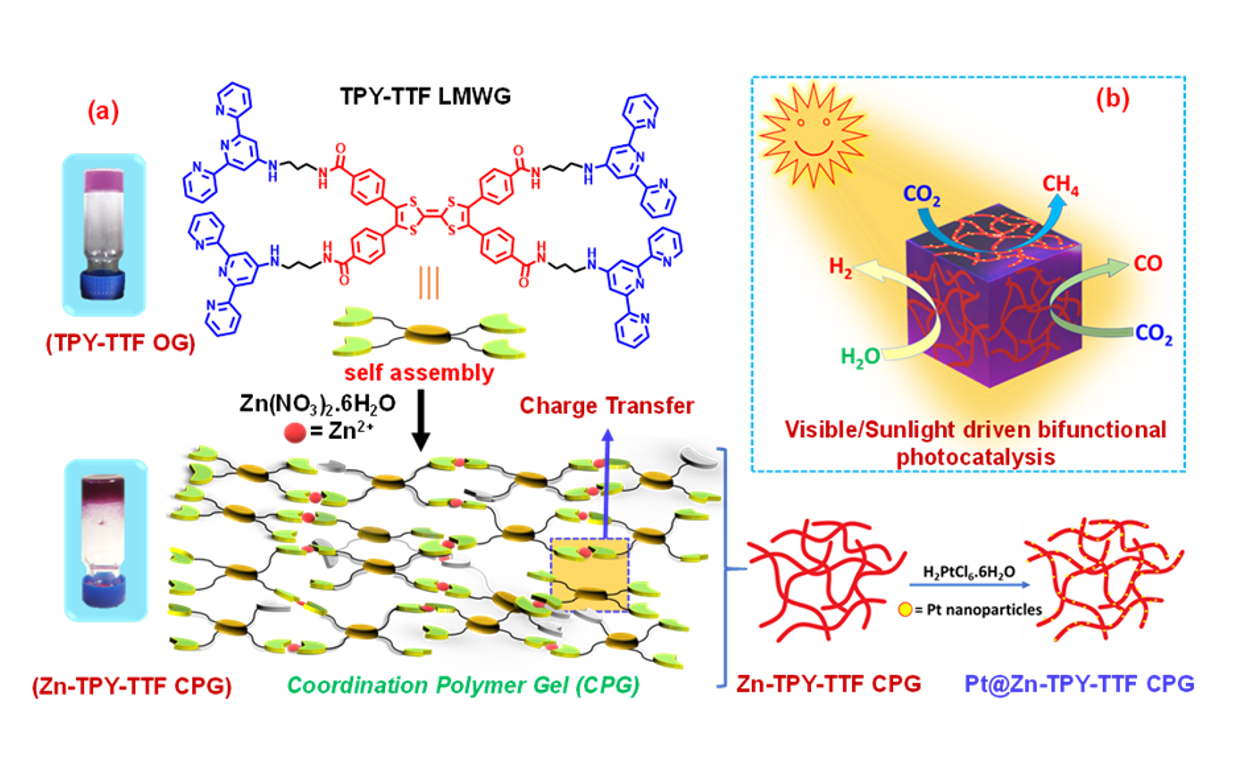
The TPY-TTF LMWG was synthesized by the amide coupling reaction between 2,2¢:6¢,2²-terpyridin-4¢-yl-propane-1,3-diamine (TPY-NH2)54 and 1,3,6,8-tetrakis (benzoic acid) tetrathiafulvalene (TTF(COOH)4)55 (details are given in supporting information (SI)), Supplementary Fig. 1-2). The newly synthesized TPY-TTF LMWG was characterized by NMR, Mass, FT-IR (Supplementary Figs. 3-6). UV-Vis absorption study was performed for a well-characterized TPY-TTF LMWG in methanol. 10-6 M methanolic solution of TPY-TTF showed distinguished absorption bands at 270 nm and 320 nm corresponding to the π®π* transition for TPY unit and TTF core, respectively (Fig. 1c). Notably, a low energy absorption band appeared at 520 nm that can be ascribed to intramolecular charge transfer (CT) interaction between TTF core and benzo-amide moiety.55 The CT property of TPY-TTF has also been supported by density functional theoretical (DFT) computation where highest occupied molecular orbital (HOMO), and lowest unoccupied molecular orbital (LUMO) is centered in TTF and benzo-amide groups, respectively (Fig. 3b).
Next, we have examined gelation propensity of TPY-TTF LMWG in several solvent compositions (see the SI). Notably, heating the solution of TPY-TTF in MeOH/DCM/H2O (2:1:1) at 60 °C followed by cooling to room temperature (rt) resulted in a purple coloured opaque gel (TPY-TTF OG) (Supplementary Figs. 7-8). Further, the formation of the gel was confirmed by the inversion test method (Fig. 1a). Morphology of the TPY-TTF xerogel was recorded by the Atomic Force Microscopy (AFM) and Field Emission Scanning Electron Microscopy (FE-SEM) that showed micron size staked layered type of morphology (Fig. 1e-1f). Transmission Electron Microscopy (TEM) images have further confirmed such morphologies (Fig. 1g). Powder X-ray diffraction (PXRD) study of the TPY-TTF xerogel showed a peak at 2q =24° with a d-spacing of 3.7 Å, indicating that the self-assembly was driven by intermolecular π-π stacking interactions (Fig. 1d). Further, high-resolution TEM image showed the ordering with the lattice fringes at 3.7 Å, also suggesting π-π interactions formed 2D stacking of layers (Fig. 1b, 1g: inset). The UV-Vis absorption study for TPY-TTF xerogel displayed slightly red-shifted absorption as compared to the methanolic solution of TPY-TTF LWMG (Fig. 1c). It showed absorption bands at 300 nm and 330 nm, which can be assigned for π®π* transitions of terpyridine and TTF units, respectively. Notably, a broad absorption between 520-560 nm was also observed, indicating the existence of CT in TPY-TTF in xerogel state. The optical band gap for TPY-TTF OG calculated by the Kubelka-Munk plot derived from UV–vis diffuse reflectance spectrometry was found to be 2.26 eV (Supplementary Fig. 9). The experimental band gap was further supported by performing density functional theory calculations on simplified models of TPY-TTF OG systems (Fig. 3a). The TD-DFT calculations have indicated that the intramolecular charge transfer transition took place at wavelength 540 nm in TPY-TTF OG from donor TTF core to acceptor -PhCONH2 moiety (Fig. 3b and Supplementary Fig. 10). The intermolecular distance between TTF and terpyridine moiety in the TPY-TTF OG was estimated theoretically and found to be 3.558 Å which indicates for the feasibility for the intermolecular CT as well (Fig. 3a). The computed results suggested the intramolecular charge transfer contribute much significantly (47%) compared to the weak intermolecular charge transfer process (1.3%). Thus, the results revealed that the visible light absorption in the TPY-TTF OG is mainly attributed to the intramolecular CT rather than intermolecular CT process. Next, the presence of four terpyridine units in the TPY-TTF gelator has prompted us to investigate further their metal- binding ability to develop coordination polymer gel (CPG) for widening their applications. To this end, we have chosen ZnII as a metal node for binding with TPY as such self-assembly is ubiquitously being explored due to soft acid-base interaction. We have performed titration of TPY-TTF (8 ×10-6 M in MeOH) with a methanolic solution of Zn(NO3)2.6H2O (8 ×10-4 M), and corresponding UV-Vis absorption spectra were recorded (Fig. 2b). Notably, the presence of isosbestic point in the ZnII titration suggested the complex formation between ZnII and TPY-TTF LMWG.54 Job’s plot analysis carried out using UV-Vis titration data illustrates the binding ratio of Zn(NO3)2 and TPY-TTF is likely to be 2:1. Next, Zn(NO3)2 and TPY-TTF gelator was taken in a molar ratio of 2:1 in the solvent mixture of MeOH/DCM/H2O (2:1:1). Heating the reaction mixture to 60 °C followed by cooling to room temperature has afforded a deep purple coloured coordination polymer gel (Zn-TPY-TTF CPG) (Fig. 2a, Supplementary Figs. 11–14). FESEM analysis of Zn-TPY- TTF showed entangled nanofibrous like morphology, suggesting metal coordination drastically changed the morphology in CPG (Fig. 2h). The length and diameter of nano-fibres were found to be between 0.1 µm – 1 µm and 40-80 nm, respectively. Further, AFM images of Zn-TPY-TTF showed the height nanofibers was found to be ~7 nm (Figs. 2e-2g). The elemental mapping of the xerogel exhibited the uniform distribution of ZnII in a 3D network of the CPG (Supplementary Fig. 13). EDAX and elemental analyses also correlated the 2:1 ratio of Zn: TPY-TTF in the CPG (Supplementary Fig. 12). TEM studies also revealed similar morphologies as observed in FESEM and high-resolution TEM analysis exhibited ordering in the nanofibres and lattice fringes were observed with the distance of 3.6 Å which could be attributed to the intermolecular π-π stacking between the TTF and TPY from the [Zn(terpy)2] units (Fig. 2i-2j and 3a). Further, the PXRD pattern of Zn-TPY-TTF CPG showed a peak at 2θ = 24.9° (3.6 Å), justifying the presence of π-π stacking (Fig. 2d). The UV-Vis absorption spectrum of Zn-TPY-TTF in xerogel state was found to be similar to the TPY-TTF OG with a small bathochromic shift in the absorbance (+10 nm) as shown in Fig. 2c. The optical band gap for Zn-TPY-TTF was calculated to be 2.27 eV which is closer to the bandgap of TPY-TTF OG (Supplementary Fig. 9). Notably, TD-DFT calculations performed on Zn-TPY-TTF demonstrates a weak intramolecular CT (3%) at 568 nm (Supplementary Fig. 15b). At the same time, a strong intermolecular charge transfer (41%) from TTF core to terpyridine unit was observed at the same wavelength (568 nm) (Supplementary Fig. 15a) with additional intermolecular CT band at 588 nm (Fig. 3b). Intermolecular CT interactions could occur via π-π stacking, as supported by lattice fringes observed in the HR-TEM images. Importantly, terpyridine units in Zn-TPY-TTF were found to be more electron-deficient in nature and seem to be better electron acceptor as compared in TPY-TTF OG (Fig. 3b). Next, as a controlled study, we found energies of the LUMO for TTF(CONH2)4 and [Zn(terpy)2] are -1.90 eV and -1.92 eV, respectively (Fig. 3c), which indicates that excited-state electron transfer is energetically favourable from TTF(CONH2)4 core to [Zn(terpy)2] centre.
Next, Mott Schottky (MS) analysis was performed for the xerogel of both, TPY-TTF and Zn-TPY-TTF to evaluate experimental feasibility for water and CO2 reduction (see SI for details). The MS plots exhibited n-type nature with a positive slope for both TPY-TTF and Zn-TPY-TTF (Fig. 4a). The flat band potentials (Vfb) were found to be -0.60 V and -0.54 V versus RHE (at pH=7) for TPY-TTF and Zn-TPY-TTF, respectively. Based on the bandgaps obtained using UV–vis diffuse reflectance spectrometry (Supplementary Fig. 9), the electronic band structures versus RHE at pH 7 could be elucidated and are displayed in Fig. 4b.56 Interestingly, the band alignments are shown in Fig. 4b illustrate that both TPY-TTF and Zn-TPY-TTF possess suitable band edge positions to perform water and CO2 reduction under visible-light irradiation.
Photocatalytic activity under laboratory condition:
(a) Visible light driven photocatalytic H2 production from water:
We have examined the potential of CPG for photocatalytic H2 production from water under visible light (400–750 nm) irradiation using 300 W xenon lamp as the light source (experimental details are given in the SI). Photocatalytic activity of Zn-TPY-TTF was examined in both, gel and xerogel state, and similar H2 evolution was observed for both under similar conditions (Supplementary Fig. 16a). However, catalytic activities in different conditions were performed in the xerogel state because of ease handling of the catalyst in comparison to the gel state. After optimizing the catalyst loading (Supplementary Figs. 17), 1 mg of Zn-TPY-TTF in xerogel state was dispersed in 38 ml of water for the photocatalytic H2 production, and 2 ml triethylamine (TEA) was added into it that acted as a sacrificial electron donor. The photocatalytic activity of Zn-TPY-TTF was monitored by gas chromatography (GC) analysis and showed 10.60 mmol/g of H2 evolution in 20 h (activity = ~530 μmolg-1h-1) upon visible light irradiation. The amount of H2 evolved was reached to saturation in 20 h and turnover number (TON) was calculated to be 23.5 (see SI) as shown in Fig. 4e. The activity is quite impressive and indeed higher than many other transition metal-based photocatalysts (Supplementary Tables 3-5). The Q.E. for H2 production using Zn-TPY-TTF catalyst was calculated to be 0.76 % at 550 nm. Further, the absence of H2 formation with Zn-TPY-TTF under dark condition (absence of light) confirming light is an essential component for the catalysis. Catalytic recyclability/reusability and stability are important factors to evaluate overall photocatalytic performance. To examine the recyclability, the photocatalytic study was performed with Zn-TPY-TTF for 6 h, and corresponding H2 evolution was monitored (Supplementary Fig. 16b). After the first cycle, the reaction mixture was purged with N2 gas for 30 minutes, and complete removal of the H2 was ensured by the GC-analysis. Again, this reaction mixture was reused for photocatalysis, and this process was continued back and forth for four consecutive cycles. Interestingly, the amount of H2 evolution was found to be similar in every cycle, confirming the excellent recyclability of the photocatalyst. Next, recycled catalyst (Zn-TPY-TTF) was analysed by FE-SEM and TEM studies and suggested no significant change in the structure and morphology after the catalytic reaction, indicating high stability of the catalyst (Supplementary Figs. 16c-d).
Next, we have examined photocatalytic activity for the OG to compare the importance of morphology, i.e., the spatial arrangement of the chromophore and also the role of metal directed assembly in CPG. Experimental conditions employed for the TPY-TTF OG was similar to the CPG. Interestingly, the H2 evolution by the TPY-TTF OG upon visible light irradiation was increased exponentially and reached saturation in 22 h (Fig. 4e). The maximum H2 evolution in 22h was calculated to be 2 mmol/g (activity~100 μmol g-1h-1), which is albeit lesser than the CPG but higher than the most of reported metal-free photocatalysts for H2 evolution.57
To understand the significant difference in photocatalytic activities between TPY-TTF OG and Zn-TPY-TTF CPG, photocurrent measurements were performed for both (Fig. 4d) in the presence and absence of light. The photocurrent for Zn-TPY-TTF in the presence of light was found to be double as compared to TPY-TTF OG. This indicates the facile charge-separation in Zn-TPY-TTF under light irradiation; therefore, expected to show better photocatalytic activity as compared to TPY-TTF. This argument has further validated by the EIS measurement, where the charge transfer resistance for Zn-TPY-TTF was observed to be significantly lesser as compared to TPY-TTF under both, dark and light irradiated conditions (Fig. 4c). This can be attributed to the 1D nanofibril morphology of Zn-TPY-TTF CPG that provides a continuous charge transfer pathway (via co-facial intermolecular charge delocalization) for the photogenerated electrons which ultimately enhances the photocatalytic activity.
We have also performed the photocatalytic study with individual structural units of Zn-TPY-TTF as control experiments to evaluate the importance of coordination assembly of CPG in the catalysis (Supplementary Fig. 18). Notably, individual components such as TEA, TTF(COOH)4 and TPY-TTF were found to be incapable of catalysing H2 evolution reaction. Next, the photocatalytic study has also been performed by making a blend of ZnII salt with TPY-TTF (in 2:1 ratio) (details are given in SI). This showed aggregated spherical morphology as confirmed by FE-SEM study (Supplementary Fig. 19) and corresponding TON of H2 evolution calculated for 6 h was found to be three-times lesser as compared to the CPG (Supplementary Fig. 18). This unambiguously signify the impact of nano-structuring in the photocatalytic performances. To validate the role of intermolecular CT interaction, we have also synthesized a coordination polymer of ZnII (Zn-CP) with TTF(COOH)4 and characterized by FESEM, EDAX, elemental mapping, TGA, PXRD and UV-vis absorption study (Supplementary Figs. 20-23). Zn-CP showed micron-sized spherical particles with diameter in the range of 2-3 µm. The Zn-CP showed 0.8 mmol/g of H2 production from water in 12 h which is eight times lesser in activity in comparison to Zn-TPY-TTF CPG photocatalyst under a similar condition. Overall, control experiments have unambiguously indicated that the coordination driven spatial arrangement of donor-acceptor charge transfer interaction has a high significance in visible light photocatalytic performances of CPG (Zn-TPY-TTF).
Further, we envisioned that the entangled hierarchical fibrous structure of the coordination polymer gels could easily immobilize co-catalyst like Pt on the surface58, which would facilitate the separation of photogenerated charge carriers by decreasing diffusion length and eventually enhance the photocatalytic activity.59 Thus, we have successfully executed in-situ generation and stabilization of platinum (Pt) nanoparticles in the 3D fibrous network of Zn-TPY-TTF (Pt@Zn-TPY-TTF) (Fig. 5a, see the details in SI). High-resolution TEM and FE-SEM analysis have confirmed for the stabilization of Pt nanoparticles in fibrous networks of Zn-TPY-TTF within the size range of 2-3 nm (Fig. 5b, Supplementary Fig. 24a). Lattice fringes were observed for Pt NPs with the d-spacing value of 0.23 nm, indicating the presence of Pt (111) planes (Fig. 5c). Inductively coupled plasma optical emission spectroscopy (ICP-OES) measurement and EDAX analysis indicated the presence of ~2.7 wt% Pt in Pt@Zn-TPY-TTF composite (Supplementary Fig. 24b). The elemental mapping was ensured for the uniform distribution of Pt NPs in the gel matrix (Supplementary Fig. 25). Next, photocatalytic activity towards water reduction was examined for Pt@Zn-TPY-TTF under similar condition as employed for CPG as well as OG. Interestingly, Pt@Zn-TPY-TTF has shown remarkably enhanced catalytic activity under visible light irradiation and hydrogen evolution was calculated to be 162.42 mmol/g in only 11 h (activity=~14727 μmol g-1h-1) and corresponding TON value was found to be 360.1 (Fig. 5d). Drastically increased H2 evolution after Pt nanoparticles stabilization could be ascribed to the efficient charge separation in Pt@Zn-TPY-TTF as Pt-centre is well-known electron acceptor that accumulate a pool of electrons and subsequently exhibits efficient water reduction. Thus, the gel matrix of Zn-TPY-TTF is primarily acting as a photosensitizer for the generation of photoexcited electrons which transferred to Pt nanoparticles to reduce water into H2 using the harvested light energy. Furthermore, recyclability of Pt@Zn-TPY-TTF was evaluated in a continuous photocatalytic experiment performed for 24 h with intermittent H2 evacuation by purging N2 gas for 30 minutes in the time interval of 6 h (Supplementary Fig. 26b). This indicated for excellent catalytic recyclability as the catalytic performance after every 6 h was found to be almost similar. The Mott-Schottky analysis for Pt@Zn-TPY-TTF reveals that the conduction band edge occurs at -0.51 V vs. RHE at pH 7, which is lesser compared to the Zn-TPY-TTF CPG (-0.54 V) catalyst (Supplementary Fig. 27a). However, approximately four-folds higher photocurrent was observed for Pt@Zn-TPY-TTF as compared to Zn-TPY-TTF (Supplementary Fig. 27c), which further corroborated the facile electron transfer from the Zn-TPY-TTF to the Pt centre. The formation of Schottky junction in Pt@Zn-TPY-TTF was helpful to separate the photogenerated electron-hole pairs. This argument was further validated by the EIS measurement, where the charge transfer resistance for Pt@Zn-TPY-TTF was found to be almost half as compared to Zn-TPY-TTF under both, dark and visible light irradiation (Supplementary Fig. 27b). Further, the photoluminescence (PL) spectra of Zn-TPY-TTF showed weak emission with a maximum at 581 nm (lex = 520 nm) (Supplementary Fig. 28). PL spectra for Pt@Zn-TPY-TTF upon excitation at 520 nm showed significantly quenched emission as compared to Zn-TPY-TTF. Therefore, to gain more insight into the advantages of Pt NPs in the increased charge separation the time-resolved photoluminescence (TRPL) decay was studied on the Zn-TPY-TTF and Pt@Zn-TPY-TTF, as shown in Supplementary Fig. 29. A higher average lifetime Zn-TPY-TTF CPG system (1.95 ns) compared to Pt@Zn-TPY-TTF system (0.22 ns) obtained by the TRPL studies confirms that migration of photoexcited electrons is much faster in Pt@Zn-TPY-TTF compared to Zn-TPY-TTF.60,61 The decrease in the lifetime is attributed to the enhanced separation and transfer efficiency of photogenerated electrons in Pt@Zn-TPY-TTF system, which plays decisive roles in enhancing the efficiency of photocatalytic processes. Furthermore, quantum efficiency (Q.E.) for the water reduction to H2 was determined for the Pt@Zn-TPY-TTF upon irradiating with monochromatic light of the wavelength of 400 nm, 450 nm, 500 nm, 550 nm, 600 nm, 650 nm and 700 nm (Supplementary Fig. 30). Notably, the highest Q.E. was obtained to be 14.47 % at 550 nm. This experiment is evident that the photocatalytic activity is mainly driven through intermolecular charge transfer interaction. The H2 evolution using Pt@Zn-TPY-TTF was examined under both light and dark conditions (Supplementary Fig. S26c). No H2 evolution was detected under dark condition, indicating the importance of light for the water reduction. Next, the photocatalysis was also performed for Pt@Zn-TPY-TTF and Zn-TPY-TTF CPG without any sacrificial donor (TEA). The aqueous dispersion of Zn-TPY-TTF produced 0.92 mmol/g of H2 in 20 h and which is 11 times lesser than with TEA. Similarly, Pt@Zn-TPY-TTF showed 30 times lesser activity without TEA (Supplementary Table 3).
(b) Visible light driven photocatalytic CO2 reduction:
As mentioned above, the theoretical and experimental bandgap alignment of Zn-TPY-TTF CPG and TPY-TPY OG is in good agreement to reduce the CO2 as well. Therefore, we have performed visible-light-driven photocatalytic CO2 reduction studies with the xerogel of Zn-TPY-TTF and also compared with TPY-TTF OG. TEA was used as a sacrificial electron donor (SD) for CO2 reduction (details are given in SI). First, screening of the solvent composition for CO2 reduction has been performed (Supplementary Figs. 31-32) and mixture of acetonitrile: water (3:1) have shown the best activity. Visible light driven CO2 reduction by Zn-TPY-TTF yielded 3.51 mmol/g of CO in 8 h with > 99% selectivity (activity= ~438 μmol g-1 h-1). The Q.E. of CO2 photoreduction for Zn-TPY-TTF CPG at 550 nm was calculated to be 1.71%. Such an impressive CO formation with outstanding selectivity is noteworthy and one of the best results among various reported hybrid photocatalysts systems (Supplementary Tables 6-8). The TON for Zn-TPY-TTF in 8 h was calculated to be 7.8 (Fig. 6b). The stability of Zn-TPY-TTF CPG was examined in the time interval of 6 h up to four cycles and found intact as for the first cycle, indicating the excellent catalytic stability (Supplementary Fig. 39a). Further, the photocatalytic activity of Zn-TPY-TTF was examined upon isotopic labelling with 13CO2 (see SI). This showed the formation of 13CO, which confirms that the produced CO was originated from CO2 (Supplementary Figs. 33-34). Next, visible light driven photocatalytic CO2 reduction have also investigated for OG (TPY-TTF) under a similar condition as employed for Zn-TPY-TTF (Fig. 6a). The TPY-TTF has displayed 1.12 mmol/g CO formation in 11 h with >99% selectivity (activity= ~140 μmol g-1 h-1) and corresponding TON was estimated to be 2.1 (Fig. 6a).
We have performed in situ Diffuse Reflectance Infrared Fourier Transform (DRIFT) spectroscopic study for Zn-TPY-TTF CPG to track the reaction intermediates formed in the course of CO2 reduction to CO (see details in SI).56 Two peaks appeared at 1514 and 1692 cm-1 were assigned for the COOH* and COO-* intermediates, respectively, which are the signature intermediates forms initially during the CO2 reduction (Fig. 7a).62 Peaks observed at 1454 cm-1 could be attributed to symmetric stretching of the HCO3-*.63 In addition, a peak at 1275 cm-1 could be assigned for the C-OH* of the HCO3-* intermediate.63 A noteworthy peak at 2074 cm-1 was indicated for formation of the CO*. Most importantly, peak intensity of the CO* intermediate was substantially increased with reaction progress, suggesting CO* formation increases with time. Based on the experimental results and in-situ study, we have computed plausible mechanism for CO2 reduction which is in good agreement with earlier report as well27 (Fig. 7b). The photocatalytic cycle is initiated by the light absorption at photosensitiser, which is TTF core followed by reductive quenching of TTF* by TEA. Next, an electron generated from the TTF* was transferred to [Zn(TPY)]2+ and subsequently produced the radical cation species [Zn(TPY.-)(TPY)]1+. Notably, the electron was localized at terpyridine unit of the catalyst that resulted [Zn(TPY.-)(TPY)]1+ species with the stabilization energy of 0.36 eV (Supplementary Fig. 43a). Next, as the reaction progress, a terpyridine ligand was leaving from the coordination sphere of ZnII and eventually solvent molecules (acetonitrile) occupy the vacant coordination site to afford a complex [Zn(TPY.-)(MeCN)2]1+ (Supplementary Fig. 43b). This complex binds with a CO2 molecule and produce the [Zn(TPY)(MeCN)2(CO2-)]1+ species (Supplementary Fig. 44d). As evidenced by the DFT results, Zn-CO2 distance in the [Zn(TPY)(MeCN)2(CO2-)]1+ was decreased to 2.319 Å from 3.017 Å compared to transition state species (TS) (Supplementary Fig. 44c). Notably, CO2 molecule acts as a monodentate ligand, and the angle for ∠O-C-O was found to be 140.75o keeping in mind that free CO2 has linear geometry (Supplementary Fig. 44d). Here, it is worth mentioning that the one-electron charging over the [Zn-TPY] unit favours the binding of CO2 molecule by increasing electron density at the metal centre, which is prerequisite for the nucleophilic attack to the CO2. However, the [Zn(TPY)(MeCN)2(CO2-)]1+ complex was further reduced to form the singlet species [Zn(TPY.-)(MeCN)2(CO2-)] (Supplementary Fig. 44e). This carboxylate centre of the singlet complex reacts with the second molecule of CO2 and subsequently releasing a carbonate anion from the catalytic cycle and resulting in the formation of [Zn(TPY)(MeCN)2(CO)]2+ complex (Supplementary Fig. 44g). In this complex, the CO molecule is loosely attached to the ZnII centre at a distance of 2.585 Å. As a result, it can be easily released from the metal centre and therefore, catalyst molecule re-enters into the catalytic cycle.
Next, the photocatalytic activity of Pt@Zn-TPY-TTF towards CO2 reduction has also been examined under a similar experimental condition as applied for CPG and OG. Pt@Zn-TPY-TTF is expected to show impressive catalytic activity due to the presence of Pt NPs. And indeed, this has depicted excellent CO2 reduction and more interestingly, displayed CH4 formation rather than CO (Fig. 6d). It has already been reported in the literature that the presence of Pt NPs on the surface of semiconductor plays a key role for the formation of CH4.58 The low H2 dissociation barrier and weak bond (H-Pt) facilitate the CO2 bonding with hydrogen on the Pt surface.58,59,64 Thus, Pt NPs acts as atomic hydrogen reservoir that supplies protons readily for CH4 formation while CO2 reduction. Screening of the solvent condition for Pt@Zn-TPY-TTF indicated that the maximum conversion of CO2 to CH4 can be achieved in the mixture of acetonitrile: water (3:1) (Supplementary Fig. 32). The formation of CH4 was reached to saturation in 30 h and the corresponding yield was calculated to be 8.74 mmol/g (activity= ~292 μmol g-1 h-1) (Fig. 6d). During CO2 reduction, a small amount of H2 evolution was also observed (~0.20 mmol/g in 30 h) which was more in the initial hours but significantly decreased as the reaction progressed with time. Thus, the selectivity of CO2 reduction to CH4 formation in 30 h was noted to be more than 97 % (Fig. 6e). The TON for CH4 was calculated to be 19.4 in 30 h, which is indeed impressive (Supplementary Table 8). The catalytic performance of Pt@Zn-TPY-TTF was examined for 6 h up to four cycles, and a similar amount of CH4 formation in every cycle has ensured for excellent catalytic recyclability (Supplementary Fig. 39b). The quantum efficiency (Q.E.) of the Pt@Zn-TPY-TTF towards CO2 reduction was calculated at different wavelengths using monochromatic light (Supplementary Fig. 37). The highest Q.E. for the CH4 formation was obtained to be 0.81 % at 550 nm which further confirms that the photocatalytic activity of Pt@Zn-TPY-TTF is attributed to the intermolecular charge transfer interactions. The CO2 reduction was also performed under both, light and dark conditions using Pt@Zn-TPY-TTF (Supplementary Fig. 38). The CH4 formation was not increased under dark condition, justifying the importance of light in the photocatalysis. Further, photocatalysis was performed with labelled 13CO2 (isotopic labelling) using Pt@Zn-TPY-TTF (Supplementary Figs. 35-36). This showed the formation of labelled 13CH4 and confirming that CO2 is the actual source for the CH4 formation. Next, we have performed DRIFT experiment for Pt@Zn-TPY-TTF in order to monitor the real-time progress for the CO2 reduction reaction (Fig. 8). Similar to the Zn-TPY-TTF, IR-stretching peaks observed for Pt@Zn-TPY-TTF at 1610 cm-1, 1570 cm-1 and 1426 cm-1 could be attributed to the intermediates COOH*, COO* and HCO3*, respectively.56 Weak intensity peak for the CO* at 2062 cm-1 illustrated that the CO* could be readily converted to other multi-electron reduction intermediates. Moreover, the characteristic intermediates for CH4 formation were observed at 1081 cm-1 (CHO*), 1015 and 1120 cm-1 (CH3O*).56 This clearly showed that the presence of Pt NPs in the Pt@Zn-TPY-TTF CPG play catalytic role towards modulating the CO2 reduction product from CO to CH4.
Sunlight driven photocatalytic studies:
(a) Sunlight driven H2 evolution:
Above discussions have clearly shown that visible-light photocatalytic activity and stability of both, Zn-TPY-TTF and Pt@Zn-TPY-TTF in the xerogel state is indeed impressive. Notably, the amount of H2 evolution obtained using photocatalyst Zn-TPY-TTF is higher than previously reported non-precious metal-based photocatalyst materials (Supplementary Table 5). Therefore, our next goal was to examine the potential of Zn-TPY-TTF towards H2 evolution upon direct sunlight irradiation under ambient condition (Supplementary Fig. 40). Nevertheless, we have performed photocatalysis experiment with Zn-TPY-TTF in sunlight from 9:00 am to 4:00 pm for one week during 12th April 2019 – 17th April 2019 at the rooftop of our institute. The weather condition corresponding to the above-mentioned period can easily be found out on the web. Interestingly, maximum H2 evolution of 5.14 mmol/g in 6 h (activity= ~857 μmol g-1 h-1) upon direct sunlight irradiation was observed on 15th April which is quite impressive and indeed comparable with the amount of H2 obtained under laboratory conditions (Xe-lamp irradiation). The TON for H2 evolution was calculated for the above-mentioned period (Fig. 4f). The highest TON value of 11.9 was obtained on 15th April 2019. Whereas, the lowest TON was calculated to be 7.2 on 12th April 2019 because of partially cloudy weather. To the best of our knowledge, this work demonstrates the first example for the direct sunlight driven impressive H2 evolution exploiting non-precious metal derived soft photocatalyst material.
Next, similar to Zn-TPY-TTF, we have also examined sunlight driven photocatalytic H2 evolution for Pt@Zn-TPY-TTF composite in xerogel state (Fig. 5e). The experimental condition for Pt@Zn-TPY-TTF was similar to the Zn-TPY-TTF. Nevertheless, experimental timing was different for the Pt@Zn-TPY-TTF. The sunlight driven photocatalysis with Pt@Zn-TPY-TTF was performed from 27th June 2019 to 29th June 2019. Interestingly, water reduction efficiency of Pt@Zn-TPY-TTF under sunlight irradiation was extremely high. The highest H2 evolution was calculated to be 72 mmol/g in 6 h (activity= ~12000 μmol g-1 h-1) on 27th June 2019, and corresponding TON was calculated to be 159.7 (Fig. 5e). Whereas, the lowest TON value for H2 evolution was found to be 114.6 on 28th June due to partially cloudy weather.
(b) Sunlight driven CO2 reduction:
Interestingly, the potential of Zn-TPY-TTF for CO2 reduction has also been examined under direct sunlight between 9:00 a.m. to 4:00 pm for three days from 28th to 30th Sept 2019 (Fig. 6c). The highest CO formation of 1.79 mmol/g was observed in 6 h (activity= ~298 μmol g-1 h-1) on 30th September 2019, and corresponding TON was calculated to be 3.9 (Fig. 6c). The CO formation under direct sunlight irradiation is albeit lower than the laboratory condition but the obtained amount under ambient condition is quite exciting and promising because of practical application.
Next, sunlight driven CO2 reduction has been performed with Pt@Zn-TPY-TTF for three days from 1st October 2019 to 3rd October 2019 (Fig. 6f). Similar to the laboratory conditions, Pt@Zn-TPY-TTF composite upon sunlight irradiation has displayed CH4 formation as the results of CO2 reduction. The highest CH4 formation of 0.96 mmol/g in 6 h (activity= ~160 μmol g-1 h-1) was observed on 1st October 2019 and corresponding TON value was calculated to be 2.1. Whereas, the lowest CH4 evolution with the TON value of 1.9 took place on 2nd October 2019. Furthermore, after performing sunlight driven photocatalysis with Zn-TPY-TTF CPG, the catalyst was recovered through centrifugation and washed with fresh water 3-4 times. The FE-SEM and TEM analysis performed for recovered catalyst sample has confirmed that the fibrous morphology of the Zn-TPY-TTF remained intact, whereas EDAX and elemental analysis ensured the presence of all the elements in similar quantity as obtained for as-synthesized Zn-TPY-TTF (Supplementary Fig. 41). This unambiguously demonstrates the excellent stability of the Zn-TPY-TTF CPG during sunlight irradiation. Similarly, the structural integrity of the recovered Pt@Zn-TPY-TTF after sunlight driven photocatalysis was analysed by microscopic techniques. FE-SEM & TEM images of recovered Pt@Zn-TPY-TTF was found to be similar to the as-synthesized material, indicating for excellent stability of the catalyst during photocatalysis (Supplementary Fig. 42).
{kind=link}