Estrogen deficient osteocytes produce soluble factors which induce osteoclastogenesis and bone resorption. However, inhibiting sclerostin can revert these effects.
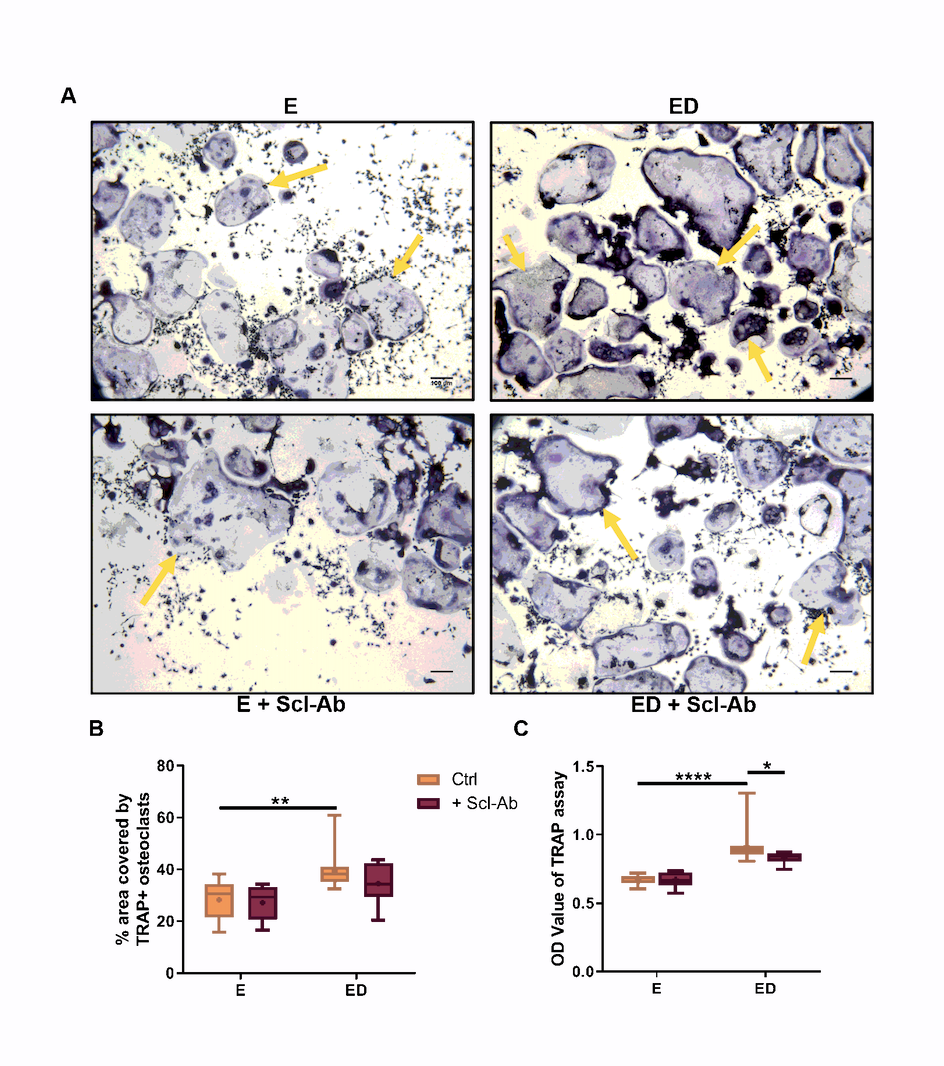
In vitro, osteocytes that have undergone an estrogen withdrawal regime have been shown to have impaired mechanosensation and altered pro-osteoclastogenic mRNA expression (RANKL/OPG, COX-2) [23]. To mimic the conditions observed in estrogen deficiency, osteocytes were subjected to an estrogen withdrawal regime, in which estrogen was withdrawn from the cells that had been accustomed to estrogen for 3 days. Following oscillatory fluid flow, CM was collected and CM experiments were conducted to investigate the paracrine signalling between mechanically stimulated osteocytes and osteoclast precursors. There was significantly higher TRAP + cells when BMMs were cultured with CM from estrogen deficient (ED) osteocytes (p < 0.01) and also higher levels of TRAP activity (p < 0.0001) compared to CM from estrogen treated osteocytes (E) (Fig. 1A,C,D). Bovine bone discs were used to assess the resorptive ability of the differentiated osteoclasts and a significant increase in resorption was observed when BMM cells were cultured with CM from estrogen deficient osteocytes compared to CM from estrogen treated osteocytes (p < 0.0001) (Fig. 1B,E). An increase in osteoclastogenesis and TRAP activity was also observed when RAW264.7 cells were treated with CM from estrogen deficient osteocytes compared to estrogen treated osteocytes (Supplementary Fig. 1).
Scl-Ab administration in vivo has shown to prevent bone loss induced by mechanical unloading and ovariectomy in a rat model of osteoporosis [4]. Here we examined whether administration of Scl-Ab to estrogen deficient osteocytes in vitro could reduce the increase in osteoclastogenesis observed when BMMs were exposed to CM from estrogen deficient osteocytes. When estrogen-treated osteocytes were treated with Scl-Ab, they produced a CM that decreased osteoclastogenesis (p < 0.05) and TRAP activity (p < 0.0001) in BMMs further, when compared to estrogen-treated cells that received no Scl-Ab (Fig. 1A, C, D). Similarly, there was a significant decrease in osteoclastogenesis (p < 0.01) and TRAP activity (p < 0.0001) when BMMs were treated with CM from estrogen deficient osteocytes treated with the Scl-Ab when compared to CM from untreated estrogen deficient osteocytes (Fig. 1A, C, D). Bone resorption by BMM was significantly reduced (p < 0.0001) when receiving CM from osteocytes exposed to Scl-Ab, when compared to groups receiving CM from estrogen-treated or estrogen deficient osteocytes with no antibody (Fig. 1B, E). In addition, a two-way ANOVA revealed a significant interaction between the inhibitory effects of estrogen and Scl-Ab on bone resorption (p = 0.0002; Fig. 1E). However, analysis by two-way ANOVA revealed no significant interaction between the effects of estrogen and Scl-Ab on osteoclastogenesis or TRAP activity. Increased osteoclastogenesis and TRAP activity was also observed when RAW264.7 cells were treated with CM from estrogen deficient osteocytes compared to estrogen treated osteocytes. However, CM collected from osteocytes exposed to Scl-Ab did not have the same inhibitory effect on RAW264.7, which was observed in BMM cell cultures (Supplementary Fig. 1).
In summary, estrogen deficient and mechanically loaded osteocytes produce soluble factors that increase osteoclastogenesis, TRAP activity and bone resorption when compared to estrogen treated and mechanically loaded osteocytes. However, inhibiting sclerostin reduces pro-osteoclastogenic paracrine signalling between osteocytes and osteoclast pre-cursors under both estrogen and estrogen deficient conditions.
Estrogen deficient osteocytes treated with Scl-Ab produce soluble factors which downregulate CTSK and NFATc1 expression in osteoclasts
NFATc1 is master regulator of osteoclast differentiation, and regulates a number of osteoclast specific gene such as TRAP and CTSK [43]. CTSK transcript encodes for cathepsin K, a protease which breaks down type I collagen and therefore plays an important role in bone resorption [44]. We assessed the effects osteocytes CM had on both CTSK and NFATc1 expression in BMM cells. BMM cells upregulated the expression of NFATc1 (p < 0.05) following 5 days of culture with CM from estrogen deficient osteocytes compared to CM from estrogen-treated cells but not CTSK expression (p = 0.09) and (Fig. 2A, B). Supporting the downregulation in bone resorption observed, the CM from osteocytes exposed to Scl-Ab significantly downregulated BMM CTSK expression when compared to the relevant CM from either estrogen-treated or estrogen deficient osteocytes that received no antibody (p < 0.01 and p < 0.05 respectively) (Fig. 2A). No significant difference in NFATc1 expression was observed when BMM cells were treated with CM from estrogen and Scl-Ab treated osteocytes compared to CM from osteocytes treated with estrogen only. However, a downregulation in NFATc1 expression was observed when BMM cells were treated with CM from Scl-Ab treated estrogen deficient osteocytes compared to CM from untreated estrogen deficient osteocytes (p < 0.01) (Fig. 2B). Analysis by two-way ANOVA revealed no significant interaction between the effects of estrogen and Scl-Ab on NFATc1 and CTSK expression. Taken together, estrogen deficient osteocytes treated with Scl-Ab produce soluble factors that downregulate the expression of genes necessary for normal osteoclast formation and function.
Estrogen deficient osteocytes that were mechanically stimulated induce osteoclastogenesis in a direct cell-cell contact co-culture system, but in the presence of Scl-Ab osteoclastogenesis is attenuated
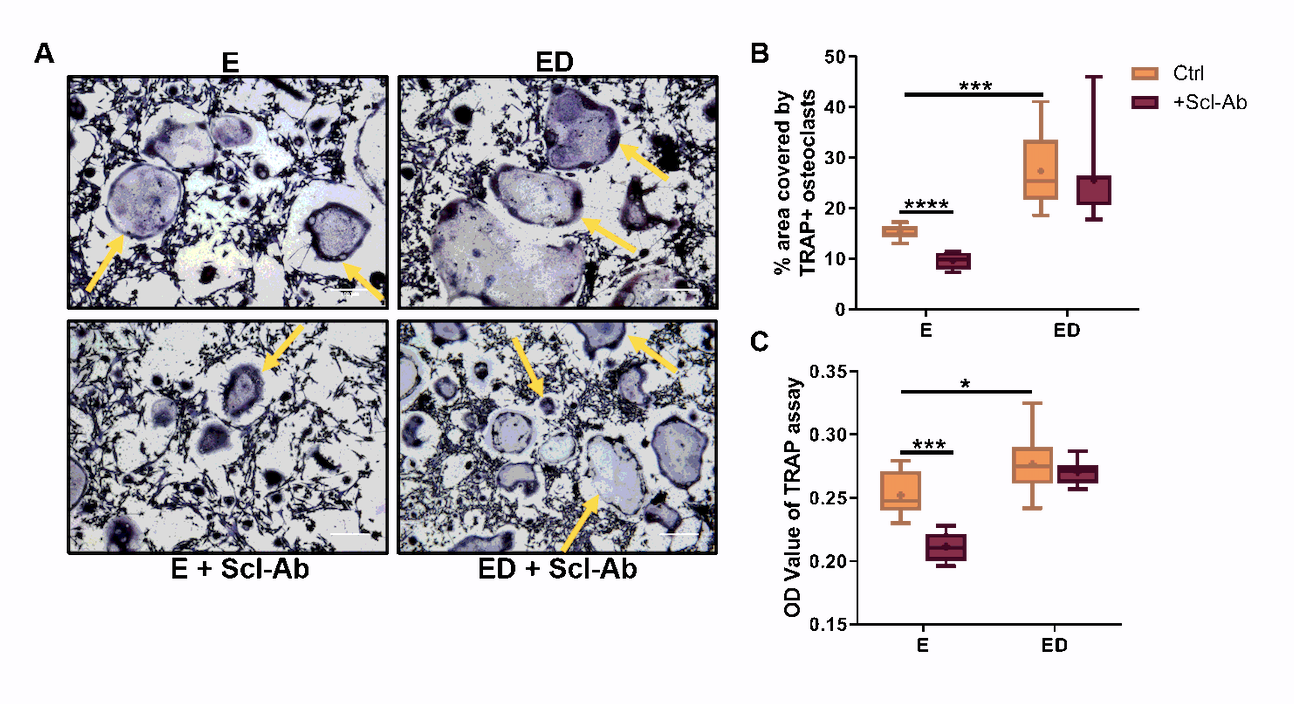
Cell-cell co-culture studies have previously revealed that mechanically stimulated MLO-Y4 cells inhibit osteoclastogenesis in both mouse BMMs and RAW264.7 cells [21, 22]. However, it is unknown what effect estrogen deficiency has on this process, and for this reason we conducted co-culture studies in which BMM cells were seeded on top of mechanically stimulated osteocytes that had previously received either estrogen treatment or an estrogen withdrawal regime. We report that estrogen deficient osteocytes had an enhanced capacity to induce osteoclast differentiation in BMM cells when compared to estrogen-treated cells (p < 0.05) and a concomitant increase in TRAP activity (p < 0.0001) was also seen (Fig. 3A, B, C). Co-cultures with RAW264.7 cells produced similar results (Supplementary Fig. 2).
We then assessed the effects of inhibiting sclerostin on osteocyte-osteoclast cell-cell signalling. In estrogen treated osteocytes, Scl-Ab treatment decreased osteoclastogenesis in BMMs (p < 0.05) but had no significant effect on TRAP activity (Fig. 3B, C). Similar results were seen when estrogen treated osteocytes were co-cultured with RAW264.7 cells (Supplementary Fig. 2). Administration of Scl-Ab reduced osteoclastogenesis (p < 0.05) and TRAP activity (p < 0.0001) when BMMs were co-cultured with estrogen deficient osteocytes and indeed these reverted to levels comparable with estrogen-treated co-cultures (Fig. 3B, C). Similar to the CM experiments, analysis by two-way ANOVA revealed no significant interaction between the effects of estrogen and Scl-Ab on osteoclastogenesis or TRAP activity in the co-culture systems.
In summary, mechanically stimulated estrogen deficient osteocytes have an enhanced capacity to induce osteoclastogenesis in a BMM co-culture system, when compared to estrogen-treated osteocytes which have also received mechanical stimulation. However, when estrogen deficient osteocytes are co-cultured with BMM cells in the presence of Scl-Ab, osteoclastogenesis is attenuated.
RANKL/OPG ratio is increased in estrogen deficient osteocytes, however, RANKL/OPG ratio is reduced following Scl-Ab administration reduces osteoclastogenesis and TRAP activity in a co-culture system
Osteocytes are the main source of RANKL, a cytokine that induces osteoclastogenesis [45]. We assessed whether the increases in osteoclastogenesis when BMMs cells were co-cultured with estrogen deficient osteocytes, or it’s CM, could be explained by changes in RANKL or OPG expression levels. There was no statistically significant difference in RANKL or OPG expression levels between estrogen deficient and estrogen-treated osteocytes (Fig. 4A, B). The RANKL/OPG ratio increased significantly in estrogen deficient cells compared to estrogen treated osteocytes (p < 0.01) (Fig. 4C).
Scl-Ab altered the RANKL/OPG ratio in estrogen-treated osteocytes; there was a significant down-regulation in RANKL expression (p < 0.01) and an up-regulation in OPG expression (p < 0.01), resulting in a decrease in RANKL/OPG ratio (p < 0.0001) compared to estrogen-treated osteocytes that received no Scl-Ab (Fig. 4A, B, C). Analysis by two-way ANOVA revealed a significant interaction between the effect of estrogen and the effect of Scl-Ab administration on the expression of these genes (RANKL: p = 0.0194, OPG: p = 0.0012 and RANKL/OPG: p = 0.0118; Fig. 4). In estrogen deficient osteocytes, Scl-Ab did not alter RANKL or OPG expression individually (Fig. 4A, B). However, the RANKL/OPG ratio was decreased in estrogen deficient osteocytes treated with Scl-Ab compared estrogen deficient osteocytes who received no Scl-Ab treatment (p < 0.05) (Fig. 4C).
Taken together, these results demonstrate that estrogen withdrawal leads to a decrease in RANKL/OPG ratio expression compared to estrogen treated osteocytes. However, administration of Scl-Ab to estrogen deficient osteocytes is capable of reducing RANKL/OPG ratio expression.
Upregulation of CXCL12 an osteoclastogenic regulatory gene in estrogen deficient osteocytes is reversed by Scl-Ab
Sclerostin inhibition can regulate expression of osteoclast regulators independent of the RANKL-OPG pathway [38]. For this reason, we analysed the mRNA expression of four secreted osteoclastogenic regulatory genes to determine the effect of estrogen deficiency and sclerostin inhibition on their expression. CXCL12 and CXCL14 expression were upregulated in estrogen deficient osteocytes when compared to estrogen treated osteocytes (p < 0.05 and p < 0.01 respectively) (Fig. 5A, B) but there was no difference in WISP1 and DLX5 expression between estrogen deficient and estrogen treated osteocytes (Fig. 5C, D). In estrogen deficient osteocytes, Scl-Ab downregulated CXCL12 expression (p < 0.05) (Fig. 5A). WISP1 expression was upregulated when estrogen treated and estrogen deficient osteocytes were administered with Scl-Ab, when compared to untreated groups (p < 0.01 and p = 0.05) (Fig. 5C). However, Scl-Ab had no effect on DLX5 expression in either estrogen treated or estrogen deficient osteocytes (Fig. 5D). Analysis by two-way ANOVA did not detect a significant interaction between the effects of estrogen and Scl-Ab on the expression of these osteoclastogenic regulatory genes.
Estrogen deficiency causes downregulation in the Wnt antagonists FRZB, SFRP2 and WIF1 but upregulates SOST expression in osteocytes
The canonical Wnt signalling pathway is an important regulator of bone homeostasis. There are a number secreted Wnt antagonists, which can be categorised as those that bind to Wnt ligands and those that bind to the Wnt co-receptor LRP5/6 [32, 33]. We analysed the mRNA expression of a number of Wnt antagonists including; WIF1, those belonging to the soluble frizzled-related protein family; SFRP2 and FRZB, as well the gene that encodes for sclerostin; SOST. Under estrogen deficient conditions there was a downregulation in WIF1 (p < 0.05) and FRZB (p < 0.05) expression compared to estrogen treated osteocytes (Fig. 6A, C). SOST expression was upregulated in estrogen deficient osteocytes compared to estrogen treated osteocytes (p < 0.05) (Fig. 6D). Administration of Scl-Ab to estrogen-treated osteocytes downregulated expression of WIF1 (p < 0.05) and SFRP2 (p < 0.05) but had no effect on FRZB or SOST expression compared estrogen-treated osteocytes that received no Scl-Ab (Fig. 6). However, Scl-Ab had no significant effect of Wnt antagonist expression when administered to estrogen deficient osteocytes (Fig. 6). Analysis by two-way ANOVA did not detect a significant interaction between the effects of estrogen and Scl-Ab on the expression of these Wnt antagonists.
{kind=link}
{kind=link}
{kind=link}